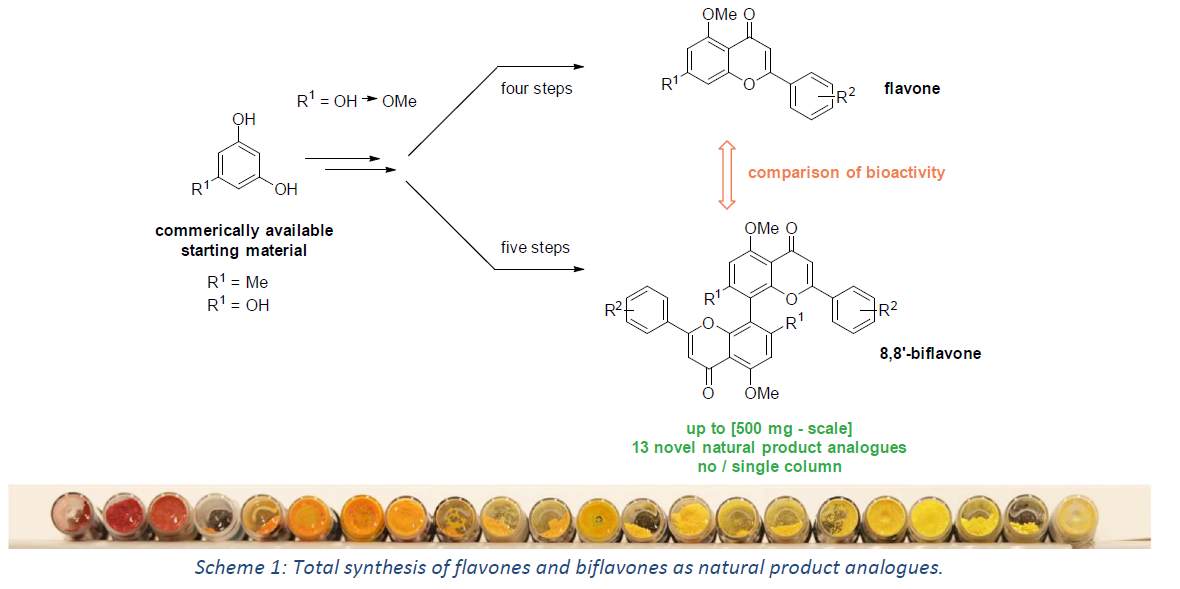
The Scientific Organizing Committee of the ACS Publications Symposium: Biological and Medicinal Chemistry accepted 90 abstracts, which were presented during the symposium in Bonn, Germany from March 6-8, 2023.
Five poster winners and one fan favorite were selected based on their exceptional research. We caught up with each of the winners to learn about what inspires their work, their mentors, and important unsolved issues in their respective fields.
Zaida L. Almeida, University of Coimbra
Smith B. Babiaka, University of Tübingen
Alica Fischle, University of Münster
Dr. Anna Junker, University of Münster
Dr. Antoine L. D. Wallabregue, University of Oxford
The Scientific Organizing Committee of the ACS Publications Symposium: Biological and Medicinal Chemistry accepted 90 abstracts to present in Bonn, Germany. Poster Presentations will take place Tuesday and Wednesday in Aula of the main building at the University of Bonn. Posters marked T will present Tuesday and marked W will present Wednesday. Presenters may check with the registration desk if they are unsure which day they are presenting. Attendees may also browse the posters in-between sessions and breaks.
Poster Session 1: Tuesday, March 7 12:40-14:10 CET
Poster Session 2: Wednesday, March 8 12:10-13:40 CET
Use the alphabetically links below to jump to each author and view their entire abstracts, highlights, and acknowledgements. Questions? Contact us at symposium@acs.org
"Development of new treatment for Neurodegenerative diseases"
We present a new approach to the capture and detection of protein aggregates using synthetic chemical antibodies to develop new diagnostics tools of the neurodegenerative diseases.
Development of new treatment for Neurodegenerative diseases
Hadia Almahli* and Christopher A. Hunter* *Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, United Kingdom
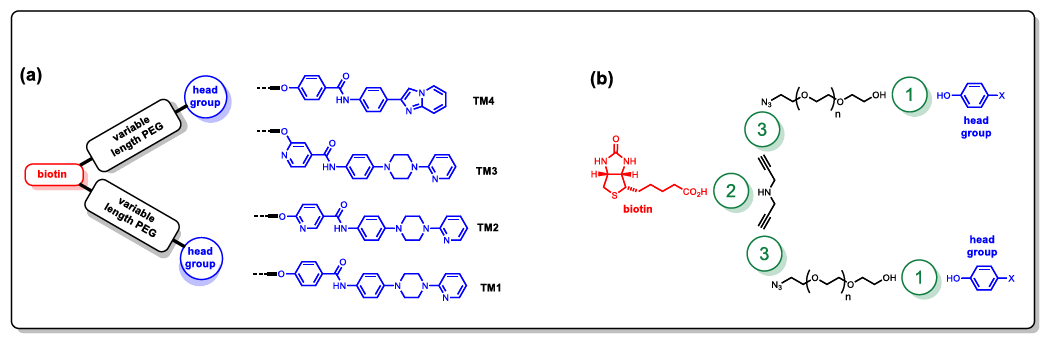
A key process in the development of neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases is the aggregation of proteins to produce fibrillary aggregates with a cross β-sheet structure, amyloid1.2. The development of reagents that can bind these aggregates with high affinity and selectivity has potential for early disease diagnosis. We describe a new approach to the capture and detection of protein aggregates using synthetic chemical antibodies. The concept is to pulldown all the aggregates present based on their structure, in this case β -sheet structure (amyloid), rather than their protein composition in order to identify which protein aggregates are present in human biofluids3. We synthesized new chemical antibodies consist of two similar head groups linked by variable linker (PEG) length using different types of coupling chemistry aiming to get high affinity and high selectivity for a α synuclein aggregates and to use the new reagents to pulldown α-synuclein aggregates from CSF.
References:
Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A. and Radford, S. E., Nat. Rev. Mol. Cell Biol., 2018, 19, 755–773. 3
Krebs, M. R. H., Bromley, E. H. C and Donald, A. M., J. Struct. Biol., 2005, 149, 30–37.
Sanna, E.; Rodrigues, M.; Fagan, S. G.; Klenerman, D.; Spillantini, M. G.; Aigbirhio, F. I.; Hunter, C. A. Mapping the Binding Site Topology of Amyloid Protein Aggregates using Multivalent Ligands. Chem. Sci. 2021, 12, 8892–8899
About the Author
Hadia is a research associate in the University of Cambridge interested in the development of new diagnostics tools of the neurodegenerative diseases, she is also a fellow in teaching of the higher education academy in the UK a STEM ambassador.
"Structural diversification of bioactive indole-based alkaloids using enzymatic methylation"
Enzymatic methylation could grant a solution for the general lack of selectivity and the high toxicity of chemical methylation processes, providing a greener way to harness the "magic methyl effect" in drug design. In this work, we propose an enzymatic approach for the stereoselective methylation of molecules containing the indole ring - a known privileged scaffold for bioactivity.
Structural diversification of bioactive indole-based alkaloids using enzymatic methylation.
Diana-Alexandra Amariei,a Nadiia Pozhydaeva,a Benoit David,b Pascal Schneider,a Julia Tenhaef,c Stephan Noack,c Thomas Classen,c Holger Gohlke,b,d Oliver Weiergräber,e Jörg Pietruszkaa,c
a. Institute for Bioorganic Chemistry, Heinrich Heine University Düsseldorf in Forschungszentrum Jülich; b. IBG-4: Bioinformatics, Forschungszentrum Jülich; c. IBG-1: Biotechnology, Forschungszentrum Jülich; d. Institute for Pharmaceutical and Medicinal Chemistry, Heinrich Heine University Düsseldorf; e. IBI-7: Structural Biochemistry, Forschungszentrum Jülich
The indole and pyrroloindoline structural motifs are prevalent among bioactive natural product alkaloids with various biological activities.1 AChE inhibitor physostigmine was used as the model compound for the development of a stereoselective enzymatic methylation platform for these scaffolds. A key step in the biosynthesis of physostigmine is the enzymatic C-methylation of an indole derivative, which drives the subsequent intramolecular cyclization and forms a stereogenic centre. 2 By using a chemo-enzymatic approach, it is possible to produce physostigmine derivatives in an enantiopure fashion. Expanding the substrate scope involves the optimization of the C-methyltransferase PsmD, which is able to transform a library of structurally diverse substrates. The acquired structural and mechanistic information about the enzyme allows for catalyst engineering towards the efficient production of physostigmine, as well as known and new analogues. As a result, the production of new synthetic alkaloids becomes available, possibly providing new scaffolds for drug discovery and optimization.
References
de Sa Alves, F. R.; Barreiro, E. J.; Manssour Fraga, C. A. From nature to drug discovery: the indole scaffold as a ‘privileged structure’. Mini Rev Med Chem, 2009, 9(7), 782-793.
Amariei, D. A.; Pozhydaieva, N.; David, B.; Schneider, P.; Classen, T.; Gohlke, H.; Weiergraber O. H; Pietruszka, J. Enzymatic C3-Methylation of Indoles Using Methyltransferase PsmD─ Crystal Structure, Catalytic Mechanism, and Preparative Applications. ACS Catal, 2022, 12(22), 14130-14139.
About the Author
The author studied biochemical engineering and is currently enrolled in a PhD in biochemistry at Heinrich Heine University, Düsseldorf, at the Institute of Bioorganic Chemistry.
Katrina Andrews – 3
University of Oxford
"The Development of PROTACs Targeting the Transcriptional Coactivators CREBBP and p300"
Complex PROTAC molecules targeting the paralogous proteins CREBBP and p300 were synthesised and their affect on protein expression levels in cells was assessed.
The Development of PROTACs Targeting the Transcriptional Coactivators CREBBP and p300
Katrina Andrews, Dr Pascal Heitel, Dr John Harling, Prof. Ester M. Hammond, Prof. Stuart J. Conway
The University of Oxford, GSK
katrina.andrews@chem.ox.ac.uk
Proteolysis-targeting chimeras (PROTACs) are heterobifunctional molecules comprising a ligand that binds the POI joined, via a linker, to a ligand that binds an E3 ubiquitin ligase enzyme. Upon formation of a POI:PROTAC:E3 ligase ternary complex, the E3 ligase can mediate the polyubiquitination of the POI, targeting it for degradation by the 26S proteasome. This process removes the whole POI from the cell, rather than inhibiting the function of an individual domain, allowing different aspects of protein function to be investigated.1
We are exploring the use of PROTACs to degrade the transcriptional coactivators CREBBP and p300. These proteins are multifunctional paralogues that modulate gene expression through the acetylation of target proteins and by mediating protein-protein interactions. Mutations in these paralogous have been implicated in numerous cancers.2 A series of PROTACs have been synthesised which combine a CREBBP/p300 bromodomain ligand with either cereblon- or VHL-targeting E3 ligase ligands. The structure activity relationship of the CREBBP/p300 ligand has been investigated to evaluate the optimal position for linker attachment and the PROTAC molecules investigate varying the lengths and nature of the linker. The synthesis and biological effects of these compounds will be presented.
Brand, M.; Clayton, J.; Moroglu, M.; Schiedel, M.; Picaud, S.; Bluck, J. P.; Skwarska, A.; Bolland, H.; Chan, A. K. N.; Laurin, C. M. C.; Scorah, A. R.; See, L.; Rooney, T. P. C.; Andrews, K. H.; Fedorov, O.; Perell, G.; Kalra, P.; Vinh, K. B.; Cortopassi, W. A.; Heitel, P.; Christensen, K. E.; Cooper, R. I.; Paton, R. S.; Pomerantz, W. C. K.; Biggin, P. C.; Hammond, E. M.; Filippakopoulos, P.; Conway, S. J. Controlling Intramolecular Interactions in the Design of Selective, High-Affinity Ligands for the CREBBP Bromodomain. J. Med. Chem. 2021, 64 (14), 10102–10123. https://doi.org/10.1021/acs.jmedchem.1c00348.
About the Author
Katrina is in the final year of her DPhil at the University of Oxford under the supervision of Prof Stuart J. Conway.
Bettadaiah B.K. – 5
Central Food Technological Research Institute
"Natural Materials Related Diversity Oriented Synthesis (NMRDOS) of Important Spice Nutraceuticals"
Natural Materials Related Diversity Oriented Synthesis (NMRDOS) of Important Spice Nutraceuticals
Dr. B.K. Bettadaiah, Principal Scientist, Spices and Flavour Science Department, CSIR-Central Food Technological Research Institute; Mysuru – 570020, India.
Nutraceuticals are alternative pharmaceuticals that are integral part of the diet rich in spices, herbs and medicinal plants. They provide immense health benefits due to the presence of single or mixture of active components which bring physiological benefit and provide protection against diseases. These natural materials are source for the bioactive ingredients, which exerts the pharmaceutical properties. The chemical functional groups responsible for the bioactivity arises majorly due to carbonyl groups, β-hydroxy carbonyl, activated aromatic system, phenolic groups etc. The main components possessing the said activity and chemical functional groups identified in spices and medicinal plants are curcumin, gingerols, shogaols, zerumbone, karanjin etc. The clinical usage of these natural materials is limited due to their ready disintegration at biological conditions, low bioavailability and fast systemic removal from the cells. Hence, their nutraceutical benefits are unrealistic at the in vivo. It is well recognized that, certain necessary modifications by physical and chemical methods on the active nutraceutical to improve their quality is practiced in the scientific pursuits. Hence, development of congeners utilizing the chemical functionality of natural materials is reported in various scientific publications. This method of developing an improved and bio-compatible forms of natural materials helps in the discovery of lead pharmaceutics as library synthesis. Those natural materials capable of affording various functionally applicable new forms categorized as NMRDOS components. The talk will be focused on the NMRDOS of curcumin, gingerols, shogaols, zerumbone, karanjin by the presenter’s research group.
Smith B. Babiaka – 4
University of Tübingen, Germany
"Molecular Networking of Novel Antimicrobial Leads from Euspongia sp. and Spongia sp. Marine Sponges"
The main aim of the study will be to discover new marine natural products (MNPs) from bacterial symbionts from these sponges using a rational based approach. This will involve collecting and extracting marine sponges as well as isolating symbiotic and associated bacteria from them. Next, screening of the extracts in vitro from both the sponge and from cultures of sponge-associated bacteria in order to determine their antimicrobial activities and the compounds associated with that activity. The project constitutes an opportunity for capacity building in microbial NPs research and the discovery of new antimicrobials and with a mechanism of action distinct from existing ones.
Molecular Networking of Novel Antimicrobial Leads from Euspongia sp. and Spongia sp. Marine Sponges
Smith B. Babiaka1,2*, Chambers C. Hughes2,3*, Heike BrÖtz-Oesterhelt2,3
Department of Chemistry, Faculty of Science, University of Buea, Buea, Cameroon.
Department of Microbial Bioactive Compounds, Interfaculty Institute of Microbiology and Infection Medicine, University of Tübingen, Tübingen, Germany.
Cluster of Excellence-Controlling Microbes to Fight Infections, Tübingen, Germany.
Over the past decade, antimicrobial resistance (AMR) continues to be a public threat on a global scale [1]. AMR is currently responsible for over 700,000 deaths annually around the world. The trend has been predicted to exponentially rise to above 10 million deaths annually by 2050, with an estimated economic cost of $100 trillion worldwide [2]. To combat the continual emergence of multidrug-resistant pathogens, there is a critical and constant need to discover new antimicrobial natural products. With respect to the development of new antimicrobials, the marine environment holds great promise for the discovery of novel bioactive compounds. Marine sponge-associated microorganisms have exhibited good potential in producing NPs with diverse medicinal and pharmaceutical properties [3, 4]. Thus as part of our research efforts, we intend to search for new marine natural products (MNPs) with antimicrobial activity from marine sponges collected from the west coast of Africa. The sponges were collected by hand using scuba at a depth of about 20 m off the Seme Beach in Limbe, Fako Division. The marine sponges were macerated in 2.5 L of methanol at room temperature for nine days (3 x 3 days). Filtration and concentration of the crude extracts of Euspongia sp. and Spongia sp. on a rotary evaporator led to 4. 4 g and 1.3 g dark brown extracts respectively. The crude extracts would be passed through size exclusion chromatography (Sephadex LH- 20) and semipreparative high-performance liquid chromatography (HPLC) techniques to obtain pure compounds. The antimicrobial activity of the crude extracts and pure compounds would be screened using standard procedures with some modifications in my host laboratory [5, 6, 7]. The structures of the compounds will be established using LC-qTOF-MS/MS measurements, carbon-13 nuclear magnetic resonance (NMR) spectral methods assisted by the performance of two dimensional (2D) NMR techniques and comparison with published data. The study constitutes an opportunity for capacity building in microbial natural products research and the discovery of new antimicrobial leads with a mechanism of action distinct from existing ones.
Keywords: Marine natural products, jellyfish, antimicrobial activity, mechanism of action.
References:
Farha et al: Nature Microbiol 2019, 4:565-577.
O’Neill J: HM Government, 2014.
Lam KS: Curr Opin Microbiol 2006, 9:245-251.
Newman et al: J Nat Prod 2004, 67:1216-1238.
Chen et al: ChemBioChem 2020, 21:1-10.
Patel et al: Approved Standard, Vol. 35, 10th ed.; Clinical and Laboratory Standards Institute: USA, 2015.
Brötz-Oesterhelt et al: Nat Med 2005, 11:1082-1087.
About the Author
Smith B. Babiaka is a Georg Forster Alexander von Humboldt and Georg Forster-Bayer Reseach Fellow at the Department of Microbial Bioactive Compounds, University of Tübingen, Germany under Hughe’s Research Group.
Dr Smith B. Babiaka would like to thank the Alexander von Humboldt Foundation for granting him the Georg Forster Alexander von Humboldt (Ref 3.4 – CMR – 1220727 – GF-P) and Georg Forster-Bayer Research Fellowship.
Younis Baqi – 87
Sultan Qaboos University
"Novel Phoenix dactylifera L. (date palm) fruit extracts exhibit anti-proliferation activity against human pancreatic cancer cell lines"
Novel Phoenix dactylifera L. (date palm) fruit extracts exhibit anti-proliferation activity against human pancreatic cancer cell lines
Reem A. Al Alawi1,2, Jörg D. Hoheisel2, Mohamed Saiel Saeed Alhamdani2 and Younis Baqi1,*
Department of Chemistry, Faculty of Science, Sultan Qaboos University, P.O. Box 36, Postal Code 123, Muscat, Sultanate of Oman;
Division of Functional Genome Analysis, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 580, 69120, Heidelberg, Germany; *Correspondence: baqi@squ.edu.om; Tel.: +968 24141473
Pancreatic cancer has a very poor prognosis.1 Its high resistance to chemotherapeutic drugs combined with late diagnosis and early metastasis make it a most aggressive and lethal cancer.2 Hence, there is a pivotal need to find new effective curative options. Consumption of fruits and vegetables is known to reduce the incidence of cancer.3 Dietary supplements, bioactive dietary agents, or nutraceuticals isolated from plants may exhibit beneficiary effects with simultaneously low toxicity.4 In this study, natural extracts from the date palm fruit (Phoenix dactylifera L.) were studied for their anti-proliferative activity in pancreatic cancer cell lines. They demonstrated a very good anti-proliferation performance. Among the tested extracts, an n-butanol collective fraction and an ethyl acetate sub-collective fraction exhibited the strongest anti-proliferative effect. In addition, not only tumor cells but also tumor-specific pancreatic stellate cells were affected, suggesting a more comprehensive therapeutic functioning on different cell types in a pancreatic tumor. Date fruit extracts clearly contain substances that might form a basis for new treatment modalities.
References:
Huang JH, Guo W, Liu Z. Discussion on gemcitabine combined with targeted drugs in the treatment of pancreatic cancer. World J. Gastroenterol. 2023, 29, 579–581. doi: 10.3748/wjg.v29.i3.579.
Szymoński K, Chmura Ł, Lipiec E, Adamek D. Vibrational spectroscopy – are we close to finding a solution for early pancreatic cancer diagnosis? World J. Gastroenterol. 2023, 29, 96–109. doi: 10.3748/wjg.v29.i1.96.
Scheffers FR, Boer JMA, Verschuren WMM, Verheus M, van der Schouw YT, Sluijs I, Smit HA, Wijga AH. Pure fruit juice and fruit consumption and the risk of CVD: the European Prospective Investigation into Cancer and Nutrition-Netherlands (EPIC-NL) study. Br J. Nutr. 2019, 121, 351–359. doi: 10.1017/S0007114518003380.
"Importance of Ile71 in actin on histidine methyltransferase catalysis"
Importance of Ile71 in actin on histidine methyltransferase catalysis
Nurgül Bilgin,a Laust Moesgaard,a Marijn N. Maas,a Jordi C. J. Hintzen,a Apolonia Witecka,b Jakub Drozak,b Jacob Kongsted,a and Jasmin Mecinovićaa Department of Physics, Chemistry and Pharmacy, University of Southern Denmark,b Department of Metabolic Regulation, Faculty of Biology, University of Warsaw. bilgin@sdu.dk, mecinovic@sdu.dk
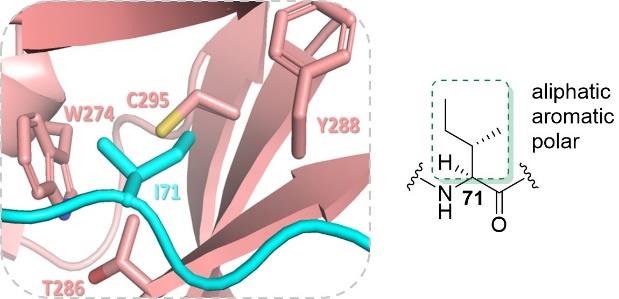
SETD3-catalysed N3-methylation of His73 in β-actin plays a key role in stabilization of actin filaments in the metazoan cells. Overexpression and/or dysregulation of SETD3 is associated with several pathologies in mammals, including cancer.1 Here, we present the role of the Ile71 residue in β-actin on human SETD3 catalysis. Incorporation of natural and unnatural Ile mimics at position 71 in β-actin peptides reveals that the ‘secondary’ I71 binding pocket of SETD3 methyltransferase modulates the substrate efficiency of β-actin. Our results demonstrates that SETD3 can accommodate structurally diverse hydrophobic side chains in its I71 pocket, providing clear limits of the size and shape of Ile analogues. Water thermodynamics calculations reveal that the Ile71 pocket is occupied by high-energy water molecules, that are released upon the Ile71 binding, contributing favourably to the SETD3-βA-SAM complex formation.2
References:
Wilkinson et al., Nature, 2019, 565, 372-376.
Bilgin et al., Org. Biomol. Chem., 2022, 20, 1723-1730.
Raitis Bobrovs – 7
Latvian Institute of Organic Synthesis
"Discovery of non-peptidomimetic plasmepsin V inhibitors"
Discovery of non-peptidomimetic plasmepsin V inhibitors
Raitis Bobrovs, Laura Drunka, Iveta Kaņepe, Aigars Jirgensons, Kristaps Jaudzems Latvian Institute of Organic Synthesis, Aizkraukles 21, Riga, LV1006, Latvia
Malaria is a deadly parasitic infectious disease that is predominantly found in tropical and subtropical areas, affecting both indigenous populations and increasing numbers of travellers. It is caused by the malaria parasite of the genus Plasmodium. Treating malaria parasite infections in the humans still depend heavily on the synthetic antimalarials, however, wide spread of drug-resistant parasite strains urges the researchers to look for drugs with a new way of action. One of the most attractive antimalarial drug targets currently is plasmepsin V (plm V) – essential malaria aspartic protease that is located in the endoplasmic reticulum. While it is structurally rather different from other Plasmodium aspartic proteases, off-target selectivity is always a great concern when developing new drugs. Due to this reason, we were interested in discovery of nonpeptidomimetic inhibitors that are known to be more selective and easier to tailor for a specific protease. Here we present non-peptidomimetic plm V inhibitors that were identified via high throughput virtual screening (HTVS) and path metadynamics (metaD) simulations, and were verified experimentally. Such inhibitors offer novel chemotypes that can be tailored to design highly selective lead-like plm V inhibitors.
Jekaterina Bolsakova – 8
Latvian Institute of Organic Synthesis
"Exploring threonyl-tRNA synthetase AMP and Zn2+ binding pocket"
The aim of our work is to identify threonyl-tRNA synthetase (ThrRS) inhibitors.
Exploring threonyl-tRNA synthetase AMP and Zn2+ binding pocket
Latvian Institute of Organic Synthesis, Riga, Latvia
jekaterina_bolsakova@osi.lv
Malaria is an infection caused by Plasmodium parasites that leads to an acute life-threatening disease. Widespread resistance of the parasite strains to clinically used antimalarial drugs drives the
researchers to discover new drugs with an unexploited mode of action.
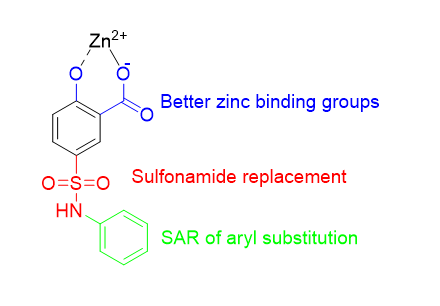
The aim of our work is to identify threonyl-tRNA synthetase (ThrRS) inhibitors. The project was started with a virtual screening of drug like compound libraries against model of ThrRS. This revealed 5-phenylsulfamoylsalicylic acid (IC50 = 13.1 µM, LE = 0.34) as a hit compound with notable structural simplicity. Based on the docking pose of the hit, its optimization was performed in three main directions (Figure 1): a) replacing phenol and carboxylic acid moiety for better zinc binding groups such as thiol and hydroxamic acid; b) sulfonamide replacement with groups making better interaction with enzymatic pocket amino acids residues, e.g. arginine; c) exploring structure-activity relationships (SAR) of aniline substitution. More than 50 hit analogues were synthesized and their activity was tested in enzymatic assay.
Figure 1. Optimization of 5-phenylsulfamoylsalicylic acid hit
About the Author
Jekaterina Bolsakova (nee Sirotkina) received her Ph. D. degree in 2020 at Riga Technical University (RTU) under Prof. Dr. chem. Aigars Jirgensons supervision.
Acknowledgements
This work was supported by the ERDF project No. 1.1.1.1/19/A/019.
Patrick J. Brennan – 9
Oxford University, Department of Chemistry
"Immunomodulatory Imide Drug Analogs Exhibit Diverse Degradation Profiles for Aiolos-Derived Zinc Finger Degron Motif"
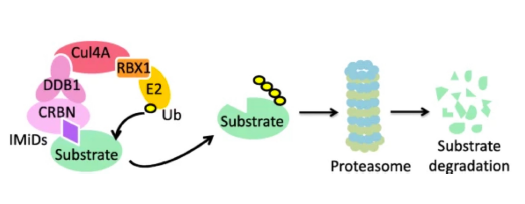
Immunomodulatory Imide Drugs (IMIDs) cause proteolytic degradation of a range of endogenous proteins in mammalian cells by recruiting them to the E3 ligase cereblon via a conserved zinc finger motif. Modification of an IMID's chemical structure leads to changes in its degradation properties.
IMMUNOMODULATORY IMIDE DRUG ANALOGS EXHIBIT DIVERSE DEGRADATION PROFILES FOR AIOLOS-DERIVED ZINC
FINGER DEGRON MOTIF
Patrick Brennan (1), Lewis Brayshaw (2), Charlotte Deane (3), Stuart Conway (1)
Chemistry Research Laboratory, University of Oxford, 12 Mansfield Rd, Oxford, OX1 3TA, United Kingdom
GSK, Gunnels Wood Rd, Stevenage, SG1 2NY, United Kingdom
Department of Statistics, University of Oxford, 24-29 St ford, OX1 3LB, United Kingdom
Immunomodulatory imide drugs (IMIDs) cause proteolytic degradation of a range of endogenous proteins in mammalian cells. In recent years, the ‘molecular glue’ mechanism by which these compounds exert their effects has been elucidated: the glutarimide ring binds to an hydrophobic pocket in the ubiquitin E3 ligase cereblon, while the phthalimide bicyclic ring alters the surface topology of cereblon. This enables the cereblon/IMID duplex to bind a family of conserved zinc finger motifs found in endogenous proteins – known as neosubstrates. Chemical modification of the phthalimide region can alter the affinity of the cereblon/IMID duplex for the neosubstrate zinc finger motif, affecting the degradation profile exhibited by the IMID in mammalian cells. Here we present a library of IMID analogues and scrutinise their degradation profiles for a zinc finger ‘degron’ derived from the neosubstrate Aiolos in a fluorescent protein assay in jurkat cells.
About the Author
Patrick J. Brennan is a PhD student in the Conway group, Oxford University. Patrick is funded by UKRI through the Synthetic Biology Centre for Doctoral Training. His research concerns molecular glues and their use in targeted protein degradation.
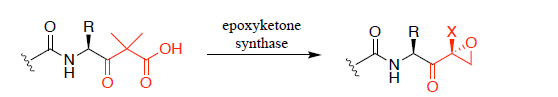
"Exploring the biosynthetic potential of epoxyketone synthases and their enzymatic mechanism"
Natural product derived epoxyketones act as potent proteasome inhibitors in cancer treatments, however their synthesis is often challenging and inefficient. This research looks to understand and exploit the biosynthesis of natural product epoxyketones to develop a chemoenzymatic and more sustainable approach for producing drug candidates.
Exploring the biosynthetic potential of epoxyketone synthases and their enzymatic
mechanism
Callum J. Bullock1, Marlene L. Rothe1,2, Joshua W. Cartwright1, Jozef R. Lewandowski1, Lona Alkhalaf1 & Gregory L. Challis1,3,4,5
Department of Chemistry, University of Warwick, Coventry CV4 7AL, United Kingdom
School of Life Sciences, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, United Kingdom
Warwick Integrative Synthetic Biology Centre, University of Warwick, Coventry CV4 7AL, United Kingdom
Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia
ARC Centre of Excellence for Innovations in Peptide and Protein Science, Monash University, Clayton, VIC 3800, Australia
Current treatments for different types of cancer include natural product derived epoxyketones,
which act as proteasome inhibitors.1 Chemical syntheses of epoxyketone drugs are highly
inefficient due to unselective formation of the epoxyketone warhead.2,3 Epoxyketone natural
products are produced by a variety of natural organisms and are biosynthesized by epoxyketone
synthases, which enantio-selectively form epoxyketones from α-dimethyl-β-keto acids (Fig.
1).4 This work aims to enable a chemoenzymatic and more sustainable method to produce
epoxyketone drug candidates. Thereby, an interdisciplinary approach was taken to investigate
epoxyketone synthases and their biosynthetic potential.
Molecular dynamics simulations of the native eponemycin substrate binding to an epoxyketone
synthase homology model revealed insights into key active site residues. The putative key
residues in EpnF, a known epoxyketone synthase, were mutated and their roles probed through in vitro assays with the mutant enzymes. Additionally, non-native substrates without several
structural features found in the natural substrate were synthesised and tested in in vitro assays
with EpnF – revealing the minimal chemical framework required for epoxyketone formation.
Further experiments will aim to investigate and expand the substrate scope of epoxyketone
synthases, to inform their full biosynthetic potential for the chemoenzymatic production of
proteasome inhibitor drugs.
Figure 1: General epoxyketone synthase catalysed reaction of α-dimethyl-β-keto acids to epoxyketones (where X = CH3, CH2OH or H).
References:
L. D. Fricker, Annu. Rev. Pharmacol. Toxicol., 2020, 60, 457–476.
H.-J. Zhou, M. A. Aujay, M. K. Bennett, M. Dajee, S. D. Demo, Y. Fang, M. N. Ho, J.
Jiang, C. J. Kirk, G. J. Laidig, E. R. Lewis, Y. Lu, T. Muchamuel, F. Parlati, E. Ring, K. D. Shenk, J. Shields, P. J. Shwonek, T. Stanton, C. M. Sun, C. Sylvain, T. M. Woo and J. Yang, J. Med. Chem., 2009, 52, 3028–3038.
J. Almaliti, P. Fajtová, A. J. O’Donoghue, M. AlHindy and W. H. Gerwick,
ChemistrySelect, 2021, 6, 12461–12465.
D. Zabala, J. W. Cartwright, D. M. Roberts, B. J. C. Law, L. Song, M. Samborskyy, P. F.
Leadlay, J. Micklefield and G. L. Challis, J. Am. Chem. Soc., 2016, 138, 4342–4345.
About the Author
University of Warwick chemistry graduate (MChem) currently working as a PhD student in the Challis group as part of the Midlands Integrative Biosciences Training Partnership (BBSRC).
"Modification of Cysteine-Substituted Antibodies Using Enzymatic Oxidative Coupling Reactions"
Modification of Cysteine-Substituted Antibodies Using Enzymatic Oxidative Coupling Reactions
Wendy Cao,a Johnathan C. Maza,a Natalia Chernyak,b John A. Flygare,b, Shane W. Krska,c F. Dean Toste,a and Matthew B. Francisa
aDepartment of Chemistry, University of California, Berkeley, California 94720, United States. bDepartment of Discovery Chemistry, Merck & Co., Inc., South San Francisco, California 94080, United States. cDepartment of Discovery Chemistry, Merck & Co., Inc., 2000 Galloping Hill Road, Kenilworth, New Jersey 07033, United States.
Cysteines are routinely used as site-specific handles to synthesize antibody-drug conjugates for targeted immunotherapy applications. Michael additions between thiols and maleimides are some of the most common methods for modifying cysteines, but these functional groups can be difficult to prepare on scale and the resulting linkages have been shown to be reversible under some physiological conditions.
Here we show that the enzyme tyrosinase, which oxidizes conveniently-accessed phenols to afford reactive ortho-quinone intermediates, can be used to attach phenolic cargo to cysteines engineered on antibody surfaces. The resulting linkages between the thiols and ortho-quinones are shown to be more resistant than maleimides to reversion under physiological conditions. Using this approach, we construct antibody conjugates bearing cytotoxic payloads, which exhibit targeted cell killing, and further demonstrate this method for the attachment of a variety of cargo to antibodies, including fluorophores and oligonucleotides.
References:
You, J.; Zhang, J.; Wang, J.; Jin, M. Cysteine-based coupling: Challenges and solutions. Bioconjugate Chem. 2021, 32, 1525–1534, DOI: 10.1021/acs.bioconjchem.1c00213
Lobba, M. J.; Fellmann, C.; Marmelstein, A. M.; Maza, J. C.; Kissman, E. N.; Robinson, S. A.; Staahl, B. T.; Urnes, C.; Lew, R. J.; Mogilevsky, C. S.; Doudna, J. A.; Francis, M. B. Site-specific bioconjugation through enzyme-catalyzed tyrosine-cysteine bond formation. ACS Cent. Sci. 2020, 6, 1564–1571, DOI: 10.1021/acscentsci.0c00940
Mogilevsky, C. S.; Lobba, M. J.; Brauer, D. D.; Marmelstein, A. M.; Maza, J. C.; Gleason, J. M.; Doudna, J. A.; Francis, M. B. Synthesis of multi-protein complexes through charge-directed sequential activation of tyrosine residues. J. Am. Chem. Soc. 2021, 143, 13538–13547, DOI: 10.1021/jacs.1c03079
Jennifer Carter – 11
University of Oxford
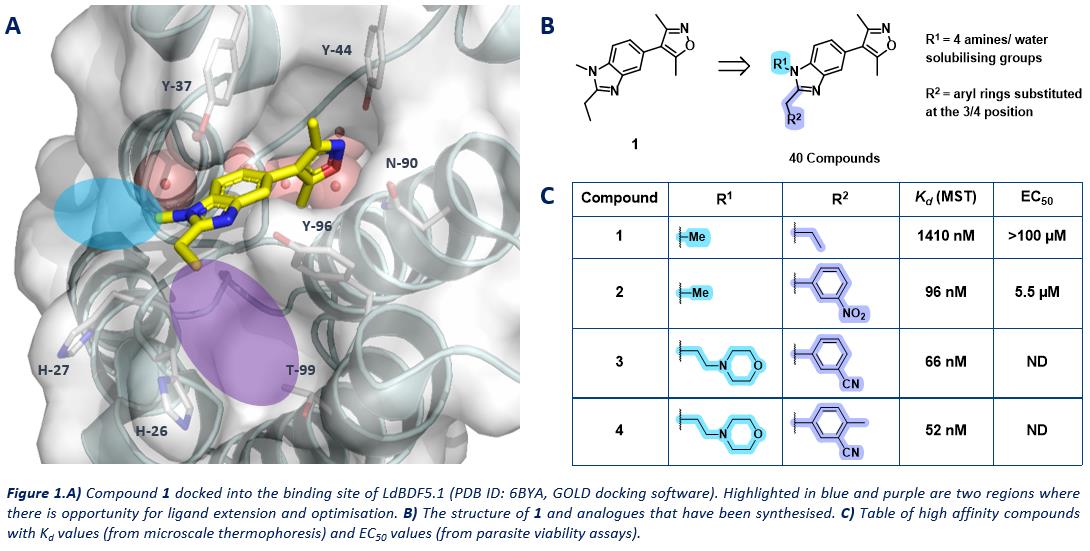
"The development of small molecule inhibitors of the essential Leishmania bromodomain LdBDF5"
We have developed high affinity ligands for LdBDF5, an epigenetic protein in Leishmania that has been genetically validated as essential to parasite survival. My work aims to chemically validate this protein as a therapeutic target to treat leishmaniasis.
The development of small molecule inhibitors of the essential Leishmania bromodomain LdBDF5
Jennifer L. Carter,1 Nathaniel G. Jones,2 Catherine Russell,2 Anthony Wilkinson,2 Jeremy C. Mottram,2 Jacob Bush,3 Stuart J. Conway1
Department of Chemistry, University of Oxford, OX1 3TA;
Department of Biology, University of York, YO10 5DD;
GlaxoSmithKline, Medicines Research Centre, Stevenage, SG1 2NY
Leishmaniasis is a neglected tropical disease caused by the parasite Leishmania. The parasites have complex lifecycles, adopting a range of phenotypes to survive in multiple hosts.1 Epigenetic processes link genotype to phenotypic diversity in a population and so must play a role in regulating the Leishmania lifecycle. Bromodomain-containing proteins (BCPs) are epigenetic proteins which recognise acetylated lysine residues in histones, and regulate transcription. LdBDF5 is a BCP in Leishmania that contains two bromodomains (LdBDF5.1 and LdBDF5.2). LdBDF5 has been genetically validated as essential for parasite survival in its two main life stages.2 The aim of this project is to develop high affinity bromodomain ligands for LdBDF5 to investigate its role in the Leishmania parasite lifecycle and chemically validate it as a therapeutic target.
A fragment-based screening approach identified compound 1 binding to LdBDF5.1 with Kd = 1410 nM (Figure 1). Analogues of 1 were synthesised with modifications at R1 and R2, to increase the affinity for LdBDF5.1. The combination of a morpholine ring at R1, and 4-methyl-3-cyanobenzene at R2 gave our highest affinity compound 4, with Kd = 52 nM. Optimised compounds for LdBDF5.1 are being tested in parasite viability assays and are demonstrating promising activity in the parasites.
References:
Field, M., Horn, D., Fairlamb, A. et al. Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat Rev Microbiol 15, 217–231 (2017)
Jones, N.G., Geoghegan, V., Moore, G. et al. Bromodomain factor 5 is an essential regulator of transcription in Leishmania. Nat Commun 13, 4071 (2022)
About the Author
Jennifer is a DPhil student in Conway group at the University of Oxford. She is working on the development of small molecule probes to investigate the role of epigenetics in the Leishmania parasite life cycle.
Subba Rao Cheekatla1 Liya Thurakkal1, and Mintu Porel 1,2*
A modular platform for the synthesis of tunable aza-oxa based macrocycles was established. Modulation in the backbone and the hanging functional groups have been rendered for achieving the tunable property. These aza-oxa based macrocycles can also differ in the number of heteroatoms in the backbone and the ring size of the macrocycles. For the proof of concept, a library of macrocycles has been synthesized with the various hanging functional groups, different combinations of heteroatoms, and the ring size in the range of 17-27 atoms in the ring. In the light of the importance of Copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction and the significance of triazole groups for various applications, we have employed the click reaction-based macrocyclization. The competence of the synthesized macrocycles in various biomedical and material applications has been proved by studying the interaction with the serum albumin protein and various heavy metals. It was observed that a few candidates, based on their hanging functional groups and specific backbone atoms, could interact well with the protein, thus improving bioactive properties. At the same time, one candidate could be used as a selective fluorometric sensor for Fe(III) ions. On the whole, this work is a proof-of-concept to explore the backbone and sidechain tunable macrocycle for different properties and applications.
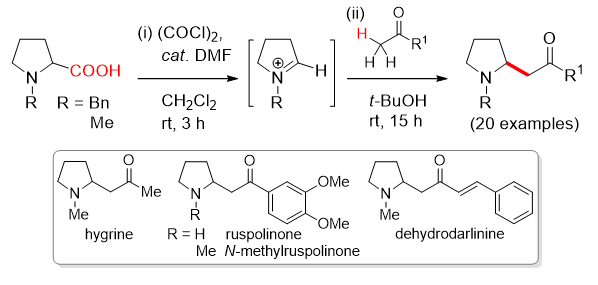
Tun-Cheng Chien
National Taiwan Normal University
"Bio-inspired Total Synthesis of Pyrrolidine and Indolizidine Alkaloids"
Bio-inspired Total Synthesis of Pyrrolidine and Indolizidine Alkaloids
Tun-Cheng Chien*
Department of Chemistry, National Taiwan Normal University
No.88, Ting-Zhou Road, Taipei 11677, Taiwan
E-mail: tcchien@ntnu.edu.tw
2-(Acylmethylene)pyrrolidine derivatives were synthesized via intermolecular decarbonylative Mannich reaction from various methyl ketones and 1-alkyl-1-pyrroliniums, generated in situ from 1-alkylprolines. This methodology features the advantages that direct formation of pyrrolinium intermediates from 1-alkylprolines and subsequent intermolecular Mannich reactions with methyl ketones could both be carried out under simple and mild conditions without the use of metal catalysts and other additives. This approach mimics the biosynthetic pathway and provides a direct access to a series of 2-(acylmethylene)-pyrrolidine alkaloids, including hygrine, N-methylruspolinone, dehydrodarlinine and ruspolinone. Meanwhile, the decarbonylative Mannich reaction was also applicable to π-electron-excess heteroarenes as the nucleophilic counterparts. A series of pyrrole and indole derivatives underwent the decarbonylative Mannich reaction with 1-alkylprolines under the same condition to give the corresponding 1-alkyl-2-heteroarylpyrrolidines in very good yields. The reactions took place exclusively at the C2-position of pyrroles and C3-position of indoles. Further application of this methodology would be amenable to the synthesis of versatile 2-substituted pyrrolidine derivatives.
"Epigenetic machinery misregulation by lncRNAs contribute to pancreatic cancer resistance to gemcitabine"
Epigenetic machinery misregulation by lncRNAs contribute to pancreatic cancer resistance to gemcitabine.
Authors: Ricardo Alberto Chiong Zevallos; Eduardo Moraes Rego Reis
Affiliations: Instituto de Química – Universidade de São Paulo
Pancreatic adenocarcinoma (PDAC) is one of the deadliest cancers and there are no defined diagnostic or prognostic biomarkers. Long non-coding RNAs (lncRNAs) are potential biomarkers and some contribute to chemoresistance against gemcitabine, the first line drug against PDAC. LncRNAs can regulate chromatin remodeling complexes such as PRC2, guiding or decoying. EZH2 is the histone methyltransferase in PRC2 that catalyzes the methylation of histone 3 at lysine 27 and its expression is misregulated in many tumors. LncRNAs can recruit EZH2 to silence tumor suppressor or activate oncogenes, independently from the PRC2 complex. Since lncRNAs misregulation in cancer might result in a differential recruitment of EZH2, it’s plausible to assume that lncRNAs might contribute to chemoresistance by guiding EZH2. The function of lncRNAs in PDAC chemoresistance was investigated by qPCR and RNA Immunoprecipitation (RIP) experiments contrasting parental and gemcitabine-resistant cells derived from AsPC-1 cell line. The enrichment of lncRNAs differentially expressed in resistant cells was assessed in the immunoprecipitated of EZH2 after RIP. The lncRNAs bound to EZH2 in chemoresistant PDAC might be responsible for the specific recruiting of EZH2 to regulate chemoresistance-associated genes. This research provides insights to new combinatorial therapies targeting EZH2 and the lncRNAs interacting with it.
References:
Alvarez-Dominguez JR, Lodish HF. Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood. 2017;130(18):1965–1975.
Bhan A, Hussain I, Ansari KI, Kasiri S, Bashyal A, Mandal SS. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol. 2013;425(19):3707-3722. doi:10.1016/j.jmb.2013.01.022
Chen P, Wan D, Zheng D, Zheng Q, Wu F, Zhi Q. Long non-coding RNA UCA1 promotes the tumorigenesis in pancreatic cancer. Biomed Pharmacother. 2016 Oct;83:1220-1226. doi: 10.1016/j.biopha.2016.08.041. Epub 2016 Aug 23. PMID: 27562722.
Duguang L, Jin H, Xiaowei Q, et al. The involvement of lncRNAs in the development and progression of pancreatic cancer. Cancer Biol Ther. 2017;18(12):927-936. doi:10.1080/15384047.2017.1385682
Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284-99.
Fu X, Deng X, Xiao W, Huang B, Yi X, Zou Y. Downregulation of NEAT1 sensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine through modulation of the miR-506-3p/ZEB2/EMT axis. Am J Cancer Res. 2021 Aug 15;11(8):3841-3856. PMID: 34522453; PMCID: PMC8414385.
Fu Z, Chen C, Zhou Q, Wang Y, Zhao Y, Zhao X, Li W, Zheng S, Ye H, Wang L, He Z, Lin Q, Li Z, Chen R. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017 Dec 1;410:68-81. doi: 10.1016/j.canlet.2017.09.019. Epub 2017 Sep 22. PMID: 28947139.
Han T, Jiao F, Hu H, et al. EZH2 promotes cell migration and invasion but not alters cell proliferation by suppressing E-cadherin, partly through association with MALAT-1 in pancreatic cancer. Oncotarget. 2016;7(10):11194-11207. doi:10.18632/oncotarget.7156
Hu, JJ., Song, W., Zhang, SD. et al. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci Rep 6, 23521 (2016)
Huarte M. The emerging role of lncRNAs in cancer. Nature medicine. 2015; 21(11):1253-1261.
Li Z, Yu D, Li H, Lv Y, Li S. Long non‑coding RNA UCA1 confers tamoxifen resistance in breast cancer endocrinotherapy through regulation of the EZH2/p21 axis and the PI3K/AKT signaling pathway. Int J Oncol. 2019 Mar;54(3):1033-1042. doi: 10.3892/ijo.2019.4679. Epub 2019 Jan 8. PMID: 30628639.
Long Y, Hwang T, Gooding AR, Goodrich KJ, Rinn JL, Cech TR. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat Genet. 2020 Sep;52(9):931-938. doi: 10.1038/s41588-020-0662-x. Epub 2020 Jul 6. PMID: 32632336.
Pawłowska E, Szczepanska J, Blasiak J. The Long Noncoding RNA HOTAIR in Breast Cancer: Does Autophagy Play a Role?. Int J Mol Sci. 2017;18(11):2317. Published 2017 Nov 3. doi:10.3390/ijms18112317
Shi Y, Wang XX, Zhuang YW, Jiang Y, Melcher K, Xu HE. Structure of the PRC2 complex and application to drug discovery. Acta Pharmacol Sin. 2017;38(7):963-976.
Wang L, Wang F, Na L, Yu J, Huang L, Meng ZQ, Chen Z, Chen H, Ming LL, Hua YQ. LncRNA AB209630 inhibits gemcitabine resistance cell proliferation by regulating PI3K/AKT signaling in pancreatic ductal adenocarcinoma. Cancer Biomark. 2018;22(1):169-174.
Wang X, Sehgal L, Jain N, Khashab T, Mathur R, Samaniego F. LncRNA MALAT1 promotes development of mantle cell lymphoma by associating with EZH2. J Transl Med. 2016 Dec 20;14(1):346. doi: 10.1186/s12967-016-1100-9. PMID: 27998273; PMCID: PMC5175387.
Wang ZQ, Cai Q, Hu L, He CY, Li JF, Quan ZW, Liu BY, Li C, Zhu ZG. Long noncoding RNA UCA1 induced by SP1 promotes cell proliferation via recruiting EZH2 and activating AKT pathway in gastric cancer. Cell Death Dis. 2017 Jun 1;8(6):e2839. doi: 10.1038/cddis.2017.143. PMID: 28569779; PMCID: PMC5520878.
Mabilly Cox Holanda de Barros Dias – 13
University of Warwick
"Initial efforts on biosynthetic engineering approaches to the chemical diversification of enacyloxin IIa."
Biosynthetic engineering of antibiotic-producing strains offers a new road towards the targeted development of new weapons against AMR. The understanding and engineering of Enacyloxin IIa production line has afforded a compound library of about 40 compounds, that will be expanded and rationalised in this current project.
Initial efforts on biosynthetic engineering approaches to the chemical diversification of enacyloxin IIa.
Mabilly Cox Holanda de Barros Dias1, Dan Van1, Lona Alkhalaf1, Greg Challis 1.
University of Warwick, Chemical Biology Research Facility (CBRF), Department of Chemistry, CV4 7SH, Coventry; mabilly.cox@warwick.ac.uk ; g.l.challis@warwick.ac.uk ; l.alkhalaf1@warwick.ac.uk
Antibiotics are essential in modern medicine, but antimicrobial resistance (AMR) is rapidly neutralising their effectiveness. Thus, an urgent need is to develop new antibiotics to overcome the health threat AMR poses. Enacyloxin IIa is an antibiotic produced by Burkholderia species with potent activity against Acinetobacter baumannii, one of the WHO “critical priority” pathogens, including antibiotic-resistant clinical isolates, that blocks protein synthesis by allosterically inhibiting EF-Tu. To optimise enacyloxin for clinical use, analogues are needed to illuminate its structure-activity relationship and improve its aqueous solubility and chemical stability. So far, a chemical library of almost 40 analogues was generated through various strategies, including bioengineering and mutasynthesis. This project aims to rationally expand the enacyloxins’ chemical space, developing structures that are stronger candidates for clinical development by having optimised drug-like properties and fragments for chemical handling. The optimisation of compound production in the native bacteria and genetic engineering experiments will also be developed to exploit bioengineering possibilities. In parallel, bioactivity assays will be conducted to test the potency of the analogues generated and expand the bioactivity range of this family of compounds.
References:
Masschelein J, Sydor PK, Hobson C, Howe R, Jones C, Roberts DM, et al. A dual transacylation mechanism for polyketide synthase chain release in enacyloxin antibiotic biosynthesis. Nature chemistry. 2019;11(10):906-12.
Mahenthiralingam E, Song L, Sass A, White J, Wilmot C, Marchbank A, et al. Enacyloxins are products of an unusual hybrid modular polyketide synthase encoded by a cryptic burkholderia ambifaria genomic island. Chemistry and Biology. 2011;18(5):665-77.
Parmeggiani A, Krab IM, Watanabe T, Nielsen RC, Dahlberg C, Nyborg J, et al. Enacyloxin IIa pinpoints a binding pocket of elongation factor Tu for development of novel antibiotics. Journal of Biological Chemistry. 2006;281(5):2893-900.
Cetin R, Rab IMK, Anborgh PH, Cool RH, Watanabe T, Sugiyama T, et al. Enacyloxin IIa, an inhibitor of protein biosynthesis that acts on elongation factor Tu and the ribosome. EMBO Journal. 1996;15(10):2604-11.
Watanabe T, Sugiyama T, Takahashi M, Shima J, Yamashita K, Izaki K, et al. The structure of enacyloxin II, a novel linear polyenic antibiotic produced by Gluconobacter sp. W-315. Agricultural and biological chemistry. 1990;54(1):259-61.
About the Author
Pharmaceutical scientist that graduated from Federal University of Pernambuco (BR) with 5 years of medicinal organic chemistry experience and 1 year in biomedical research (IE), currently a PhD candidate in Challis/Alkhalaf group (UK).
The Group is based in the Department of Chemistry at the University of Warwick and is co-led by Prof Greg Challis and Dr Lona Alkhalaf.
The main research focus of the group is the chemistry and biology of natural products e.g. antibiotics, siderophores, phytotoxins, autoinducers and other bioactive metabolites.
"Constituents from ripe figs of Ficus vallis choudae Delile (Moraceae) with antiplasmodial activity"
Constituents from ripe figs of Ficus vallis‑choudae Delile (Moraceae) with antiplasmodial activity
Ficus vallis-choudae are used in traditional medicine against several conditions including nausea and malaria. However, its use is still to be scientifically documented and validated. Hence, the aim of this work was to evaluate the antiplasmodial activity of the DCM-MeOH (1:1) crude extract, their hexane, dichloromethane (DCM), ethyl acetate, and methanol fractions, as well as the isolated constituents. The chemical study of the DCM-MeOH (1:1) extract of F. vallis-choudae figs led to the isolation of fifteen compounds identified based on their spectroscopic data and by comparison of these data with those reported in the literature. Some of the isolated compounds were assessed in vitro for their antiplasmodial activity against P. falciparum chloroquine-sensitive 3D7 (Pf3D7) and multidrug-resistant Dd2 strains. The DCM fraction exhibited very good antiplasmodial activity against both strains with IC50 values of 13.86μg/mL and 8.18 μg/mL, respectively. Among the tested compounds, wighteone was the most active against Pf3D7 (IC50=24.6±1.5 μM) and Dd2 (IC50=11.9±2.4 μM) strains. The obtained results could justify the traditional uses of F. vallis-choudae against malaria.
"Design, synthesis, in-silico and in-vitro studies- conjugates of lignin derived phenolic compounds and siderophores as Quorum sensing inhibitors via Trojan Horse Strategy for Pseudomonas aeruginosa"
Design, synthesis, in-silico and in-vitro studies- conjugates of lignin derived phenolic compounds and siderophores as Quorum sensing inhibitors via Trojan Horse Strategyfor Pseudomonas aeruginosa
Tamanna Dua a, Kusum Harjai*b and Vasundhara Singh*a
a Department of Applied Sciences, Punjab Engineering College (Deemed to be University), Chandigarh, 160012, India
b Department of Microbiology, Panjab University, Chandigarh, 160025, India
There is a tremendous increase in antibiotic resistance among the human bacterial pathogens specifically Pseudomonas aeruginosa , hence finding alternative approaches against bacterial infections is urgently needed.[1] In this regard, Quorum sensing inhibitors (QSI’s) has come into the picture and is speculated to be a trending approach to inhibit the bacterial growth.[2] Natural-oriented compounds are always taken into consideration, because they are biodegradable and usually very useful, so they serve as a convenient compound to combat the biological infection.[3] They are assumed to be better than other QSIs, so for this reason, they can be used more confidently for a prolonged time and can reach the situation of GRAS (generally recognized as safe).[4]
In the present work, The design strategy includes various conjugates of phenolic compounds derived from lignin through both non-hydrolysable and cleavable linkers. Further, it was then conjugated with siderophore mimic as potential drug delivery system called trojan horse strategy via iron acquisition pathway.[5] The synthesized conjugates were completely characterized by 1HNMR, 13CNMR, Mass and HPLC techniques. All the conjugates were analyzed with various receptors (LasR, RhlR, PqsR ) of P. aeruginosa via in-silico studies and excellent binding strengths were observed. Finally, the in vitro studies of all the conjugates were carried out and they were found to be give enhanced anti-quorum activity.
References:
P. K. Taylor, A. T. Y. Yeung, and R. E. W. Hancock, “Antibiotic resistance in Pseudomonas aeruginosa biofilms: Towards the development of novel anti-biofilm therapies,” J. Biotechnol., vol. 191, pp. 121–130, 2014, doi: 10.1016/j.jbiotec.2014.09.003.
K. Heurlier, V. Dénervaud, and D. Haas, “Impact of quorum sensing on fitness of Pseudomonas aeruginosa,” Int. J. Med. Microbiol., vol. 296, no. 2–3, pp. 93–102, 2006, doi: 10.1016/j.ijmm.2006.01.043.
+1 author D. Vattem V. Huerta, K. Mihalik, “Herbs, Spices and Medicinal Plants Used In Hispanic Traditional Medicine Can Decrease Quorum Sensing Dependent Virulence in Pseudomonas aeruginosa,” Int. J. Appl. Res. Nat. Prod., 2008.
S. A. K. S. Ahmed, M. Rudden, T. J. Smyth, J. S. G. Dooley, R. Marchant, and I. M. Banat, “Natural quorum sensing inhibitors effectively downregulate gene expression of Pseudomonas aeruginosa virulence factors,” Appl. Microbiol. Biotechnol., pp. 3521–3535, 2019, doi: 10.1007/s00253-019-09618-0.
M. Balhara et al., “Siderophores; iron scavengers: the novel & promising targets for pathogen specific antifungal therapy,” Expert Opin. Ther. Targets, vol. 20, no. 12, pp. 1477–1489, 2016, doi: 10.1080/14728222.2016.1254196.
Johanna Helen Maria Ehrler – 14
Goethe University Frankfurt
"Evaluation of selectivity and promiscuity of fatty acid mimetics"
Lipid mimetics form a group of bioactive compounds sharing pharmacological characteristics with their endogenous templates and are therefore interesting targets for medicinal chemistry.
Evaluation of selectivity and promiscuity of fatty acid mimetics
Johanna Ehrler1, Steffen Brunst2, Whitney Kilu1, Andre Krommes1, Jan Kramer1, Jan Heering2, Daniel Merk3, Eugen Proschak1
Institute of Pharmaceutical Chemistry, Goethe-University, Max-von-Laue Str.9, D-60438 Frankfurt, Germany
Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Theodor-Stern-Kai 7, D-60596 Frankfurt am Main, Germany
Department of Pharmacy, Ludwig-Maximilians-University, Butenandtstraße 5-13, D-81377 Munich, Germany
Lipids serve not only as energy source and structural element but more over as key players in signal transduction. As essential signaling molecules, they interact with a large number of proteins, including nuclear receptors, enzymes, and G-Protein-Coupled Receptors (GPCRs), making them an interesting and challenging target for drug discovery. Compounds that are structurally similar to endogenous lipid signaling molecules and bind orthosterically to their target proteins are called lipid mimetics. Especially regarding the treatment of inflammation and metabolic diseases, lipid mimetics offer a high potential since many of the lipid binding proteins have not yet been investigated or are not known and cannot be specifically addressed.1
We were able to set up a library of 159 fatty acid mimetics and tested these compounds for their selectivity and activity on a diverse set of eight proteins (FABP4, FABP5, sEH, LTA4H2, PPARγ, RXRα, FFAR1 and BLT13). 18 compounds showed an effect of at least 70% of the respective controls on targets they were not designed for and were than further evaluated by determining the respective activities.
The evaluation showed that the compounds are very selective, as most of them were only active on one of the target or dual-active.
References:
Proschak, E., Heitel, P., Kalinowsky, L. & Merk, D. Opportunities and Challenges for Fatty Acid
Mimetics in Drug Discovery. Journal of medicinal chemistry 60, 5235–5266 (2017).
Brunst, S. et al. Systematic Assessment of Fragment Identification for Multitarget Drug Design. ChemMedChem 16, 1088–1092 (2021).
Hernandez-Olmos, V. et al. First Structure-Activity Relationship Study of Potent BLT2 Agonists as Potential Wound-Healing Promoters. Journal of medicinal chemistry 63, 11548–11572 (2020).
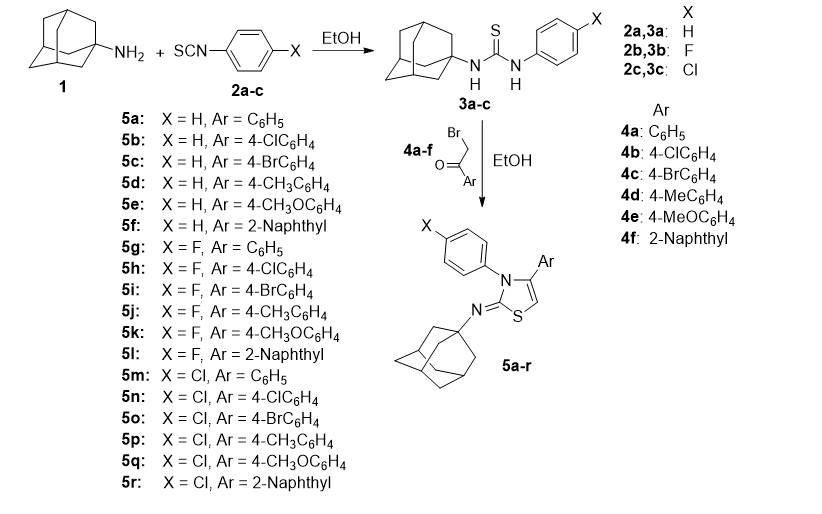
"Synthesis and in vitro antibacterial, antifungal, anti-proliferative activities of novel adamantane-linked thiazole derivatives"
Synthesis and in vitro antibacterial, antifungal, anti-proliferative activities of novel adamantane-linked thiazole derivatives
Eman T. Warda,1 Mahmoud B. El-Ashmawy,1 El-Sayed E. Habib,2 Mohammed S. Abdelbaky,3 Santiago Garcia-Granda,3 Subbiah Thamotharan,4 and Ali A. El-Emam1*
Department of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
Department of Microbiology and Immunology, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
Department of Physical and Analytical Chemistry, Faculty of Chemistry, Oviedo University-CINN, Oviedo 33006, Spain
Biomolecular Crystallography Laboratory, Department of Bioinformatics, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur-613 401, India
A series of (Z)-N-(adamantan-1-yl)-3,4-diarylthiazol-2(3H)-imines (5a-r) was synthesized via condensation of 1-(adamantan-1-yl)-3-arylthioureas (3a-c) with various aryl bromomethyl ketones (4a-f). The structures of the synthesized compounds were characterized by 1H NMR, 13C NMR and by X-ray crystallography. The in vitro inhibitory activities of the synthesized compounds were assessed against a panel of Gram-positive and Gram-negative bacteria, and pathogenic fungi. Compounds 5c, 5g, 5l, 5m, and 5q displayed potent broad-spectrum antibacterial activity, while compounds 5a and 5o showed activity against the tested Gram-positive bacteria. Compounds 5b, 5l and 5q displayed potent antifungal activity against Candida albicans. In addition, the synthesized compounds were evaluated for anti-proliferative activity towards five human tumor cell lines. The optimal anti-proliferative activity was attained by compounds 5e and 5k which showed potent inhibitory activity against all the tested cell lines. Molecular docking analysis reveals that compounds 5e and 5k can occupy the positions of NAD cofactor and the histone deacetylase inhibitor EX527 at the active site of SIRT1 enzyme.
Katharina Sophie Erlitz – 15
WWU Muenster
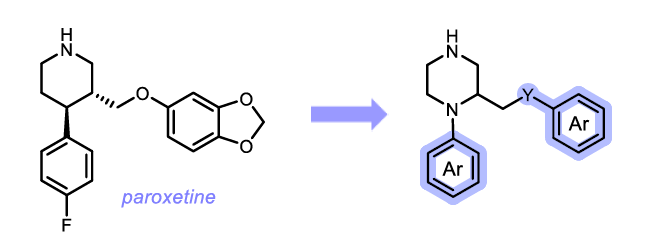
"From Paroxetine to Piperazine-based P2X4R Antagonists"
From Paroxetine to Piperazine-based P2X4R Antagonists
Erlitz, K. S.1; Junker, A.1
European Institute for Molecular Imaging (EIMI), University of Muenster, Waldeyerstrasse 15, D-48149 Muenster, Germany.
Neuropathic pain is notoriously difficult to treat; consequently, there is a high demand for novel approaches and new targets to fight this condition. The ATP-gated P2X4 receptor (P2X4R) plays an important role in the development and progression of neuropathic pain.1 There are only a few P2X4R antagonists known so far, and their allosteric binding site still remains unknown.2 The antidepressant paroxetine was demonstrated additionally to its SSRI (through serotonin transporter (SERT)) and 5-HT2C activities to inhibit the Ca2+ response in P2X4R expressing cells.3 Therefore, we believe that paroxetine serves as a perfect template for the development of novel P2X4R antagonists. A preliminary study demonstrated that the introduction of an additional N atom, leading to a piperazine ring, resulted in compounds displaying a loss of SERT and serotonin receptor activities but had no detrimental effect on P2X4R activity.4 For this reason, we pursued the strategy of exchanging the piperidine ring of paroxetine with a piperazine moiety and functionalizing its N1 position as well as the side chain with different aromatic systems to unveil SARs at P2X4Rs.
Werner S.; Mesch S.; Hillig R.C.; ter Laak A.; Klingt J.; Neagoe I.; Laux-Biehlmann A.; Dahllöf H.; Bräuer N.; Puetter V.; Nubbemeyer R.;
Schulz S.; Bairlein M.; Zollner T.M.; Steinmeyer J. Med. Chem. 2019, 62, 11194–11217.
Nagata K.; Imai T.; Yamashita T.; Tsuda M.; Tozaki-Saitoh H.; Inoue K. Molecular Pain 2009, 5, 20.
Sakuma, S.; Endo, T.; Kanakubo, N.; Arai, M.; Takahasi, T.; Imai, T.; Taguchi, K.; Nakata, E.; Mochiduki, N.; Ushioda, M.; Tsuda, M.; Inoue, K. US2011/0092703 A1.
Guilherme Fernandes – 16
University College London
"2,5-DIMETHYLPYRROLES DERIVATIVES AS POTENT AGENTS AGAINST MULTIDRUG-RESISTANT MYCOBACTERIUM TUBERCULOSIS"
Iterative medicinal chemistry campaign was performed to optimize a series of previously identified 2,5-dimethylpyrrole derivatives.
2,5-DIMETHYLPYRROLES DERIVATIVES AS POTENT AGENTS AGAINST MULTIDRUG-RESISTANT MYCOBACTERIUM TUBERCULOSIS
Guilherme F. S. Fernandes,±,ǂ Dorothy Semenya,ǂ Meir Touitou,ǂ Domiziana Masci,ǂ Camila M. Ribeiro,§ Fernando R. Pavan,§ Beatrice Gianibbi,≠ Fabrizio Manetti,≠ Daniele Castagnolo,±,ǂ
±Department of Chemistry, University College London, WC1H 0AJ, London, United Kingdom.
ǂSchool of Cancer and Pharmaceutical Sciences, King’s College London, SE1 9NH, London, United Kingdom.
§School of Pharmaceutical Sciences, São Paulo State University (UNESP), 14800-903 Araraquara, Brazil.
≠Dipartimento di Biotecnologie, Chimica e Farmacia, Dipartimento di Eccellenza 2018-2022, I-53100 Siena, Italy.
Over the past 2000 years, tuberculosis (TB) has claimed more lives than any other infectious disease. According to the last survey conducted by the WHO, in 2020 alone, the disease was responsible for 1.5 million deaths worldwide.1 Here, a series of 2,5- dimethylpyrrole derivatives were synthesized followed by in vitro characterization of their anti-Mycobacterium tuberculosis potential.2 The new series was designed from the 2,5-dimethylpyrrole scaffold of the antitubercular hit compound 1 previously identified in our group.3 The medicinal chemistry campaign has led to the discovery of new derivatives that proved to be highly selective and potent against the Mtb and MDR clinical isolates. Analogues incorporating a cyclohexanemethyl group on the methyleneamine side chain at C3 of the pyrrole core, including 2 and 3, exhibited potent inhibitory effects against Mtb strains (MIC90 of 0.73 μg/mL and 0.40 μg/mL, respectively), substantiating the essentiality of the moiety to their anti-Mtb activity. Moreover, compounds 2 and 3 demonstrated promising cytotoxicity profiles against MRC-5 and J774A.1 cell lines and proved to be effective in inhibiting the growth of intracellular mycobacteria. Further studies revealed that the new 2,5-dimethylpyrrole derivatives bind to the MmpL3, a transmembrane transporter protein involved in cell wall biosynthesis.
References:
Fernandes, G.F.S.; Thompson, A.M.; Castagnolo, D.; Denny, W.A.; Dos Santos, J.L., Tuberculosis Drug Discovery: Challenges and New Horizons. Journal of Medicinal Chemistry, 2022, 65, 7489–7531.
Semenya, D.; Touitou, M.; Masci, D.; Ribeiro, C.M.; Pavan, F.R.; Fernandes, G.F.S.; Gianibbi, B.; Manetti, F.; Castagnolo, D., Tapping into the antitubercular potential of 2,5-dimethylpyrroles: a structure- activity relationship interrogation. European Journal of Medicinal Chemistry, 2022, 237,
Touitou, F. Manetti, C. Ribeiro, F. Pavan, N. Scalacci, K. Zrebna, N. Begum, D. Semenya, A. Gupta, S. Bhakta, T. McHugh, H. Senderowitz, M. Kyriazi, and D. Castagnolo, Improving the Potency of N-Aryl- 2,5-dimethylpyrroles against Multidrug-Resistant and Intracellular Mycobacteria. ACS Medicinal Chemistry Letters, 2020, 11, 638-644.
About the Author
Currently, Guilherme is a Marie Skłodowska-Curie Research Fellow at the University College London with Dr. Daniele Castagnolo. His research interests centre on the design, synthesis, and discovery of novel antimicrobial agents, particularly anti-TB.
Institute of Food Chemistry, University of Münster
"Biologically active cyclic tetrapeptides derived from genetically modified fungi Fusarium fujikuroi"
Apicidins are natural tetrapeptides produced by filamentous fungi and known for exhibiting potent histone deacetylase inhibitory activity. By genetic manipultation and supplementation screening of notorious plant-pathogenic fungus Fusarium fujikuroi, the production of a new apicidin-like feature awaits extensive investigation including structure elucidation by high resolution mass spectrometry, nuclear magnetic resonance, and x-ray cristallography, as well as cytotoxic characterization and determination of biological activity.
Biologically active cyclic tetrapeptides derived from genetically modified fungi Fusarium fujikuroi
Alica Fischle1, Mika Lutsch1, Hans-Ulrich Humpf1, Svetlana A. Kalinina1*
Institute of Food Chemistry, Westfälische Wilhelms-Universität Münster, Germany
*Corresponding author: s_kali03@wwu.de
The fungal plant pathogen Fusarium fujikuroi has been shown to produce a variety of secondary metabolites (SMs), i.e. pigments, mycotoxins and phytohormones, some of which induce disease in crops and lead to large economic losses in agriculture every year1. However, recent activation of an SM-producing gene cluster in F. fujikuroi reported the formation of a novel apicidin-like compound, apicidin F (APF)2 (Figure 1). Genetic manipulation of this cluster lead to the production of two new apicidin-derivatives, namely apicidin J (APJ) and apicidin K (APK)2 (Figure 1). Structurally, all compounds present cyclic tetrapeptides which are potential histone deacetylase inhibitors2. These inhibitors are investigated as potential anti-tumorigenic agents, which selectively prohibit tumor cell proliferation and differentiation, ultimately inducing cell cycle arrest and cell death3,4. Furthermore, antimalarial activities have been reported for apicidin and APF2,5. Screening of the genetically modified strains of F. fujikuroi lead to the investigations of a potentially novel APF‑derivative (Figure 1). The chemical structure of this compound is studied by application of liquid chromatographic coupled to high resolution mass spectrometric techniques, nuclear magnetic resonance as well as X-ray crystallography. Additionally, putative anti‑tumorigenic activities of APF‑derivatives were tested by in silico structure‑activity investigations and further in vitro application.
Figure 1: Chemical structures of apicidins K (A), J (B), F (C) and new feature (D).
References:
Niehaus, E.-M.; Münsterkötter, M. et al., Genome biology and evolution2016, 8 (11), 3574–3599.
Niehaus, E.-M.; Janevska, S.; Bargen, K. W. von et al., PLOS ONE2014, 9 (7), e103336.
Kim, H.-J.; Bae, S.-C., American Journal of Translational Research2010, 3 (2), 166–179.
Cappellacci, L.; Perinelli, D. R. et al., Current medicinal chemistry2020, 27 (15), 2449–2493.
Darkin-Rattray, S. J.; Gurnett, A. M. et al., Proceedings of the National Academy of Sciences of the United States of America1996, 93 (23), 13143–13147.
Rita Gaidar
Chemspace
"A new AI-driven approach to hit identification"
A new AI-driven approach to hit identification
Rita S. Gaidar,1 Olga O. Tarkhanova,1 V. Joachim Haupt,2 Florian Kaiser,2 Yurii S. Moroz1
Chemspace LLC, 85 Chervonotkatska Street, Suite 1, Kyiv, 02094, Ukraine
PharmAI, Tatzberg 47, Dresden, 01307, Germany
Exploration of giga-scale chemical spaces using insilico approaches has lately been the most effective way to identify new drug candidates1,2.
The main goal of this study was to identify novel small-molecule inhibitors of Sirtuin-1 (SIRT1)3,4,5. We have utilized an AI-based6,7 method that consists of an insilico pre-selection of the compound library followed by multi-stage in vitro validation and molecular dynamics simulation to identify the best hit molecules. The selection of the focused library (434 small molecules) and analysis of SIRT1 was performed by DiscoveryEngine technology created by PharmAI.
A multistage invitro studies and validation identified nine hit compounds. One hit molecule was selected for a molecular dynamics simulation to study the mode of inhibition. Docking of the hit was applied to the substrate moiety of the reference X-ray structure of human SIRT1. The results of the study showed a hit rate of 2.1%. The approach shows a clear improvement over the known in silico methods. Application to nine hits a more rigorous label-free validation route resulted in a hit rate of 0.46%. The described approach demonstrates a hit count comparable to that of a classical high-throughput screening project while using significantly fewer compounds that must be screened.
References
Sadybekov, A. A. et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 (2022).
Müller, J. et al. Magnet for the needle in haystack: “crystal structure first” fragment hits unlock active chemical matter using targeted exploration of vast chemical spaces. J. Med. Chem. 65, 23, 15663–15678 (2022).
Douangamath, A. et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 11, 5047 (2020).
Davenport, A. M., Huber, F. M. & Hoelz, A. Structural and functional analysis of human SIRT1. J. Mol. Biol. 426, 526–541 (2014).
Chiba, S. et al. A prospective compound screening contest identified broader inhibitors for Sirtuin 1. Sci. Rep. 9, 19585 (2019).
Chan, H. C. S., Shan, H., Dahoun, T., Vogel, H. & Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 40, 801 (2019).
Gupta, R. et al. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol. Divers. 25, 1315–1360 (2021).
Shashikant Gaikwad
Shri Shivaji Mahavidyalaya, Barshi
"Study of novel Schiff base ligands and their Co(II) complexes as antitubercular, antioxidant, and anti-inflammatory agents"
Study of novel Schiff base ligands and their Co(II) complexes as antitubercular, antioxidant, and anti-inflammatory agents
Pratibha Dhale, Umesh B. Barache, Kundalkesha Gaikwad, Shashikant Gaikwad*
Chemistry Research Laboratory, Department of Chemistry, Shri Shivaji Mahavidyalaya, Barshi-413411(Maharashtra State), India
The series of new substituted 1, 3 diphenyl-1H-pyrazole-4-carbaldehydes were prepared by literature procedures. These aldehydes are then coupled with 4-amino-5-methyl-4H-1,2,4triazole-3 thiol in acidic medium to give new Schiff base of 5-methyl-4-[(Z)-3-(4-nitrophenyl)-1-phenyl-1H-pyrazol-4-yl]methylidene}amino]-4H-1,2,4-triazole-3-thiol.The synthesized ligand (MNPPMT) then treated with cobalt acetate in ethanol to form metal chelate. The synthesized ligands were characterized by IR, 1H NMR and mass spectroscopy. The metal chelate also characterized with IR, XRD and TGA to confirm the metal chelate structure. The synthesized ligand and metal chelate tested for antioxidant, antitubercular, antifungal and antibacterial activity. The study shows that the metal chelate is more active than ligands.
Manorama Ghosal
Saha Institute of Nuclear physics
"Porous Silver Core- Alloy Shell Plasmonic Nano-composite for Neuroblastoma Theranostics"
Porous Silver Core- Alloy Shell Plasmonic Nano-composite for Neuroblastoma Theranostics
M Ghosal1, C Das2, D Senapati1
Chemical Sciences Division, Saha Institute of Nuclear Physics, Kolkata, India
The emergence of nanotechnology in the field of biomedical research has attracted immense attention due to the interactions of nanoparticles (NPs) with biomolecules or bio-surfaces which lead to various emerging effects.1 Metal nanoparticles are convenient for many biophysical studies as they have localized surface plasmon resonance [LSPR] and oscillating free electrons to interact with light. Silver (Ag) has excellent LSPR properties, which can be attributed to SERS applications but Ag is highly unstable due to its rapid oxidation and high toxicity toward cells.2 The stability and biocompatibility of Ag nano-crystal can be improved by alloying with a more stable metal like Au.3 Thus, we have designed a set of core−shell nanostructures, where Au atoms are deposited on the surfaces of a sacrificial template i.e. Ag seed. These particles have high absorption and large scattering cross sections for electromagnetic radiation in the vis–NIR region thus it has crucial implications in surface-enhanced Raman spectroscopy [SERS]. In the set of particles, a good biocompatible particle has given high SERS spectra of retinoic acid, a therapeutic agent of cancer cells.4 The retinoic acid bounded core-shell nano-composites were delivered into the neuroblastoma cell as therapeutic nano-carrier for monitoring their differentiation by biochemical approaches.
References:
A. E. Nel et al.,Nature Materials,8 (2009) 543–557
J.D Padmos et al.,The Journal of Physical Chemistry C, 119(13) (2015) 7472-7482
Y. Yang et al.,ACS applied materials &interfaces,6 (2014) 3750–3757
A. Huefner et al.,Nano letters, 13 (2013)2463–2470
Alonzo Gonzalez – 18
Instituto Politécnico Nacional
"In silico evaluation of quinoxaline-1,4-di-N-oxide derivatives from ZINC15 as trypanothione reductase inhibitors"
A virtual screening of derivatives of known bioactive phenothiazine scaffold against Trypanosoma cruzi exclusive enzyme trypanothione reductase.
In silico evaluation of quinoxaline-1,4-di-N-oxide derivatives from ZINC15 as trypanothione reductase inhibitors
Instituto Politécnico Nacional, Reynosa, Tamaulipas, México
American trypanosomiasis is caused by parasite Trypanosoma cruzi, it is considered a worldwide health problem without a fully effective treatment1,2. There is thus a need to find parasite-specific alternative treatments. Trypanothione reductase (TR) is a parasite-exclusive enzyme central in parasite’s redox system crucial in detoxification of reactive species and amino acid and nucleotide biosynthesis3. Quinoxaline’s scaffold is present in molecules with trypanocidal activity4-6. A virtual screening of quinoxaline derivatives may permit finding potential TR inhibitors. TcTR crystal was obtained from the PDB database (1GXF). Ligand library of 264 compounds was compiled using substructure search of quinoxaline-1,4-di-N-oxide scaffold on ZINC15 database, 256 compounds complied with Lipinski’s and Veber’s rules and were docked in TcTR active site using AutoDock Vina 1.1.2. Binding energy was in the range of -9.6 to -5.5 kcal/mol compared to -8.8 kcal/mol of natural ligand trypanothione disulfide (TS2). Seven compounds showed a binding energy of affinity greater than or equal to natural ligand, interactions were determined for these ligands. Main interactions were found with residues important to TS2 binding: Phe396, Leu399, His461, Glu466 and Glu467. Best ranked compounds both by binding energy and interactions may be proposed as TcTR inhibitors and assayed in vitro to test effectivity.
Briceno, L.; Mosca, W. Quello Che Non Si Cerca Difficilmente Si Trova: La Malattia Di Chagas. doi.org 2016, 17 (2016Maggio), 343–347. https://doi.org/10.1714/2252.24258.
Cardoso, M. S.; Reis-Cunha, J. L.; Bartholomeu, D. C. Evasion of the Immune Response by Trypanosoma Cruzi during Acute Infection. Front Immunol 2016, 6, 659. https://doi.org/10.3389/fimmu.2015.00659.
Chacón-Vargas, K. F.; Nogueda-Torres, B.; Sánchez-Torres, L. E.; Suarez-Contreras, E.; Villalobos-Rocha, J. C.; Torres-Martinez, Y.; Lara-Ramirez, E. E.; Fiorani, G.; Krauth-Siegel, R. L.; Bolognesi, M. L.; Monge, A.; Rivera, G. Trypanocidal Activity of Quinoxaline 1,4 Di-N-Oxide Derivatives as Trypanothione Reductase Inhibitors. Molecules 2017, 22 (2), 220. https://doi.org/10.3390/molecules22020220.
González-González, A.; Sánchez-Sánchez, O.; Krauth-Siegel, R. L.; Bolognesi, M. L.; Gớmez-Escobedo, R.; Nogueda-Torres, B.; Vázquez-Jiménez, L. K.; Saavedra, E.; Encalada, R.; Espinoza-Hicks, J. C.; Paz-González, A. D.; Rivera, G. In Vitro and In Silico Analysis of New N-Butyl and Isobutyl Quinoxaline-7-Carboxylate 1,4-Di-N-Oxide Derivatives against Trypanosoma Cruzi as Trypanothione Reductase Inhibitors. Int J Mol Sci 2022, 23 (21), 13315. https://doi.org/10.3390/ijms232113315.
Torres, E.; Moreno-Viguri, E.; Galiano, S.; Devarapally, G.; Crawford, P. W.; Azqueta, A.; Arbillaga, L.; Varela, J.; Birriel, E.; di Maio, R.; Cerecetto, H.; González, M.; Aldana, I.; Monge, A.; Pérez-Silanes, S. Novel Quinoxaline 1,4-Di-N-Oxide Derivatives as New Potential Antichagasic Agents. Eur J Med Chem 2013, 66, 324–334. https://doi.org/10.1016/j.ejmech.2013.04.065.
About the Author
In our research group at Centro de Biotecnologia Genomica at Reynosa, Mexico one of our interests is finding potential new treatments for different diseases, mainly neglected diseases, this by taking advantage of in silico strategies.
Luis Donaldo Gonzalez Morales – 19
IPN
"In silico screening, molecular docking and in vitro biological activity of SARS-CoV-2 Spike (S) inhibitors"
Five inhibitors of Spike-ACE2-h interaction identified by virtual screening and molecular docking. Compound B-8 with antiviral potential with 48% in vitro inhibition of SARS-CoV-2 Spike protein-ACE2-h receptor interaction
In silico screening, molecular docking and in vitro biological activity of SARS-CoV-2 Spike (S) inhibitors
Laboratorio de Biotecnología Farmacéutica, Centro de Biotecnología Genómica-Instituto Politécnico Nacional, 88710 Reynosa, México
Department of Chemistry and SEEMS, University of Texas Rio Grande Valley, Edinburg, TX 78539, USA
Worldwide, SARS-CoV-2 has caused more than 600 million confirmed infections and 6.5 million deaths 1. The SARS-CoV-2 Spike (S) protein, whose function is to recognize and bind to the human ACE2 receptor, represents an attractive drug target for the development of antiviral agents 2. The aim of this work was to identify new compounds of natural origin as potential blockers of Spike/ACE2-h binding. A molecular docking analysis of the compounds was performed to calculate their binding pattern to the RBD domain of protein S using Withanone as a control inhibitor 3. In addition, a 120 ns molecular dynamics simulation of the lead compounds was performed and their ADME-Tox properties were estimated 4, 5. The selected compounds were evaluated by an ELISA-based enzymatic assay. In silico and in vitro studies led to the identification of compound B-8, which exhibited 48% inhibition of S/ACE2-h interaction at 50 µM and its binding pattern exhibited diverse interactions with key amino acid residues present in the RBD. This study revealed that naturally occurring compounds can bind to the RDB site of SARS-CoV-2, blocking its interaction with the ACE2-h receptor. Therefore, compound B-8 can be used as a template for developing new effective antiviral drugs.
References:
Pekar JE, Magee A, Parker E, Moshiri N, Izhikevich K, Havens JL, Gangavarapu K, Malpica Serrano LM, Crits-Christoph A, Matteson NL, Zeller M, Levy JI, Wang JC, Hughes S, Lee J, Park H, Park MS, Ching Zi Yan K, Lin RTP, Mat Isa MN, Noor YM, Vasylyeva TI, Garry RF, Holmes EC, Rambaut A, Suchard MA, Andersen KG, Worobey M, Wertheim JO. The molecular epidemiology of multiple zoonotic origins of SARS-CoV-2. Science. 2022 Aug 26;377(6609):960-966. doi: 10.1126/science.abp8337.
Day CJ, Bailly B, Guillon P, Dirr L, Jen FE, Spillings BL, Mak J, von Itzstein M, Haselhorst T, Jennings MP. Multidisciplinary Approaches Identify Compounds that Bind to Human ACE2 or SARS-CoV-2 Spike Protein as Candidates to Block SARS-CoV-2-ACE2 Receptor Interactions. mBio. 2021 Mar 30;12(2):e03681-20. doi: 10.1128/mBio.03681-20.
Balkrishna A, Pokhrel S, Singh H, Joshi M, Mulay VP, Haldar S, Varshney A. Withanone from Withania somnifera Attenuates SARS-CoV-2 RBD and Host ACE2 Interactions to Rescue Spike Protein Induced Pathologies in Humanized Zebrafish Model. Drug Des Devel Ther. 2021 Mar 11;15:1111-1133. doi: 10.2147/DDDT.S292805.
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1, 19-25.
Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports, 7(1), 1-13.
About the Author
I am Luis Donaldo Gonzalez Morales, I grew up in a small town El Mante, Tamaulipas, Mexico. I have a Master of Science in Genomic Biotechnology. I worked looking for inhibitors of the SARS-CoV-2 Spike (S)- ACE2-h interaction as new antiviral agents.
Stefan Gross – 20
University Münster (WWU)
"Novel Targeted Drug Delivery Systems for P2X7 Receptor Antagonists"
Novel Targeted Drug Delivery Systems for P2X7 Receptor Antagonists
Gross, S.1; Junker, A.1
European Institute for Molecular Imaging (EIMI), University of Muenster, Waldeyerstrasse 15, D-48149 Muenster, Germany.
Targeting small-molecule drugs to specific tissues or organs and thereby limiting their action to areas of interest holds the potential for improved safety as well as efficacy.1 The challenge lies in finding or modifying chemical structures to increase the affinity of the agent to the target without compromising its activity. In this project we developed chimeric drug delivery systems by attaching a bone binding affinity tag in form of the bisphosphonate alendronic acid via a polyethylene glycol linker to the subtype selective P2X7 receptor antagonist AZD9056.2 The latter compound, which advanced to stage II clinical trials for rheumatoid arthritis,3 belongs to the class of highly potent adamantane containing benzamides and is structuraly well suited for chemical derivatization. By synthesizing two derivatives of this compound—one with an alkyne-, the other with an azide—functionality we were able to introduce the bisphosphonate tag by copper catalyzed “click”-chemistry and form two novel drug delivery systems. These were evaluated in a YOPRO-1 upake assay and shown to retain antagonistic activity towards P2X7Rs in the nanomolar range. Since P2X7Rs are expressed by both bone and cancer cells4 these delivery systems are envisioned as innovative drug prototypes for metastatic breast cancer with the ability to accumulate at the sites of rapid bone turnover.
Figure 1: The rational design and synthesis of a composite drug delivery system with high bone binding affinity consisting of the P2X7 receptor antagonist AZD9056 and the bisphosphonate osteoporosis medication alendronic acid.
References:
Cole L. E.; Vargo-Gogola T.; Roeder R. K. Adv Drug Deliv Rev 2016, 99, 12-27.
Astra Zeneca AB, U.S. Patent WO 01/44170, June 21, 2001.
Keystone E. C.; Wang M. M.; Layton M.; Hollis S.; McInnes I. B.; Ann Rheum Dis 2012, 71, 1630-1635.
Adinolfi E.; Amoroso F.; Giuliani A. L. J Osteoporos 2012, 2012, 637863.
Roland Kofi Gyampoh – 21
University of Ghana
"Kintamdin, a ribosomally synthesised and post-translationally modified peptide consisting of a bis-thioether macrocyclic ring and a β-enamino acid residue isolated from Streptomyces sp. RK44"
Kintamdin is a macrocyclic peptide isolated from Streptomyces sp. RK44. It has an unprecedented chemical motif, bis-thioether crosslink (methyl-amino-bithionin (MAbi)), and a rare β-enamino acid ((Z)−3-amino-acrylic acid (Aaa) or ΔZ βAla) residue.
Kintamdin, a ribosomally synthesised and post-translationally modified peptide consisting of a bis-thioether macrocyclic ring and a β-enamino acid residue isolated from Streptomyces sp. RK44
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are structurally complex natural products with diverse bioactivities. In our search for novel bioactive specialized metabolites, we discovered kintamdin, a RiPP, from Streptomyces sp. RK44. RK44 was isolated from soil collected from underexplored riparian regions of Kintampo Waterfall, a biodiversity hotspot in the Eastern Region of Ghana. Metabolomic and genomic analyses of RK44 showed it is a prolific producer of unique secondary metabolites including RiPPs. The structure of kintamdin, which features a bis-thioether macrocyclic ring and a β-enamino acid residue, was elucidated using a combination of spectroscopic, spectrometric and genomic techniques. Investigation of its biosynthetic pathway demonstrated that it relies on four dedicated proteins: phosphotransferase KinD, Lyase KinC, kinase homolog KinH and flavoprotein KinI, which share low homologues to enzymes known in other RiPP biosynthesis. During the posttranslational modifications, KinCD is responsible for the formation of the characteristic dehydroamino acid residues including the β-enamino acid residue, followed by oxidative decarboxylation on the C-terminal Cys and subsequent cyclization to provide the bis-thioether ring moiety mediated by coordinated action of KinH and KinI. Conserved genomic investigation allowed further identification of two kintamdin-like peptides among the kin-like BGCs, suggesting the occurrence of RiPPs from actinobacteria.
Authors
Roland Gyampoh1, Shan Wang 3,8, Sixing Lin2,8, Qing Fang3, Zhou Lu3, Yingli Gao3,4, David J. Clarke 5, Kewen Wu3, Laurent Trembleau 3 , Yi Yu 2 , Kwaku Kyeremeh 1 , Bruce F. Milne 3,6 , Jioji Tabudravu 7 & Hai Deng3
Department of Chemistry, University of Ghana, P.O. Box LG56 Legon-Accra, Ghana.
Key Laboratory of Combinatorial Biosynthesis and Drug Discovery (MOE) and Hubei Province Engineering and Technology Research Centre for Fluorinated Pharmaceuticals, School of Pharmaceutical Sciences, Wuhan University, Wuhan 430071, China.
Department of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, UK.
College of Marine Life and Fisheries, Jiangsu Ocean University, Lianyungang, Jiangsu Province, China.
EastChem, School of Chemistry, University of Edinburgh, Edinburgh EH9 3FJ, UK.
CFisUC, Department of Physics, University of Coimbra, Rua Larga 3004-516 Coimbra, Portugal.
School of Natural Sciences, University of Central Lancashire, PR1 2HE Preston, England, United Kingdom.
These authors contributed equally: Shan Wang, Sixing Lin.
Arnison, P. G. et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 (2013).
Montalbán-López, M. et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 38, 130–239 (2021).
Ortega, M. A. et al. Structure and mechanism of the tRNAdependent lantibiotic dehydratase NisB. Nature 517, 509–512 (2015)
Ortiz-López, F. J. et al. Cacaoidin, first member of the new lanthidin RiPP fmily. Angew. Chem. Int. Ed. 59, 12654–12658 (2020).
Román-Hurtado, F. et al. Biosynthesis and heterologous expression of cacaoidin, the first member of the lanthidin family of RiPPs. Antibiotics (Basel) 10, 403 (2021).
Lohans, C. T. et al. Structure and biosynthesis of carnolysin, a homologue of enterococcal cytolysin with D-amino acids. J. Am. Chem. Soc. 136, 13150–13153 (2014).
Huo, L. & Van Der Donk, W. A. Discovery and characterization of bicereucin, an unusual D-amino acid-containing mixed twocomponent lantibiotic. J. Am. Chem. Soc. 138, 5254–5257 (2016).
Tabudravu, J. N. et al. Two distinct conformers of the cyclic heptapeptide phakellistatin 2 isolated from the Fijian marine sponge Stylotella aurantium. J. Org. Chem. 67, 8593–8601 (2002).
Tabudravu, J. N. et al. Conformational studies of free and Li+ complexed jasplakinolide, a cyclic depsipeptide from the Fijian marine sponge Jaspis splendens. Org. Biomol. Chem. 3, 745–749 (2005)
Di Costanzo, L. et al. Amino acid modifications for conformationally constraining naturally occurring and engineered peptide backbones: Insights from the Protein Data Bank. Biopolymers 109, e23230 (2018).
Crisma, M. et al. Flat peptides. J. Am. Chem. Soc. 121, 3272–3278 (1999).
Wipf, P. & Miller, C. P. A short, stereospecific synthesis of dihy-drooxazoles from serine and threonine derivatives. Tetrahedron Lett. 33, 907–910 (1992).
Wipf, P. & Miller, C. P. An investigation of the mitsunobu reaction in the preparation of peptide oxazolines, thiazolines, and aziridines. Tetrahedron Lett. 33, 6267–6270 (1992). 39. Kyosuke, S. et al. Synthesis and reaction of 6,7-dihydroazirino[1,2-a] thieno[2,3-d] pyridin-8-one. Chem. Lett. 9, 1389–1390 (1980).
Cotter, P. et al. Posttranslational conversion of l-serines to d-alanines is vital for optimal production and activity of the lantibiotic lacticin 3147. Proc. Nat. Acad. Sci. 102, 18584–18589 (2005)
Santi, S. et al. Flat, Cα,β -didehydroalanine foldamers with ferrocene pendants: assessing the role of α-peptide dipolar moments. ChemPlusChem 86, 723–730 (2021). 36. Xu, J. et al. A new cyclopeptide metabolite of marine gut fungus from Ligia oceanica. Nat. Prod. Res. 28, 994–997 (2014).
About the Author
Natural Products Chemist with 2 publications including one in Nature Communications. I hold MPhil & BSc (both Chemistry) from University of Ghana. Worked as a Medical Delegate at Nestle Ghana for almost 6yrs, currently open to PhD or MD/PhD offers.
"Selective Biocatalytic Methylation of Dipeptides as a tool for Natural Product Synthesis"
Methylation can boost the biological activity potential drugs, but there are limited possibilities of selective methylation using conventional chemical methods. As a solution to this problem, methyltransferases are used as natural biocatalysts in this research.
Selective Biocatalytic Methylation of Dipeptides as a tool for Natural Product Synthesis
M. Haasea, B. . Davidb, H. Gohlkeb,d, J. Pietruszkaa,c
a. HHU Düsseldorf; Institute of Bioorganic Chemistry
b. Forschungszentrum Jülich; Institute of Bio- and Geosciences, IBG-4: Bioinformatics
c. Forschungszentrum Jülich; Institute of Bio- and Geosciences IBG-1: Biotechnology
d. HHU Düsseldorf; Institute for Pharmaceutical & Medicinal Chemistry
To improve the pharmacological properties of compounds, methylation is an applied method for many potential bioactive drugs [1]. Selective methylation is with regard to regio- and stereoselectivity still challenging using conventional methods [2]. Having a look into nature, methyltransferases are used to create interesting frameworks in a selective manner. [3,4]. Within the family of pyrroloindole natural products, diketopiperazines (DKP), with a pyrroloindole motive derived from tryptophane, serve as interesting targets exhibiting numerous biological activities [5]. In this work, the methyltransferase StspM1 [6] was investigated due to substrate acceptance and its use for the diastereoselective methylation in a preparative scale.
References:
C.S. Leung et al., J. Med. Chem. 2012, 55, 4489-4500.
G.-J. Mei et al., Chem. Soc. Rev. 2021, 50, 5985-6012.
A. W. Struck et al., ChemBioChem 2012, 13, 2642-2655.
a) P. Schneider et al., Angew. Chem. Int. Ed. 2021, 60, 23412-23418; b) D. A. Amariei et al., ACS Catal. 2022, 12, 14130-14139.
A. D. Borthwick, Chem. Rev. 2012, 112, 3641-3716.
H. Li et al., Chem. Commun. 2019, 55, 8390-8393.
About the Author
I studied biochemistry in Düsseldorf and started in 2021 my PhD at the Institute for Bioorganic Chemistry (Heinrich-Heine University Düsseldorf).
Meike Höhl – 23
WWU Münster
"NIR Imaging Probes: A Phosphorous Rhodamine Dye for Optical Voltage Sensing"
NIR Imaging Probes: A Phosphorous Rhodamine Dye for Optical Voltage Sensing
Höhl, M. 1; Junker, A. 1
European Institute for molecular Imaging (EIMI), University of Muenster, Waldeyerstrasse 15, D-48149 Muenster, Germany
NIR-absorbing phosphorous-substituted rhodamine dyes (POR) were shown to have outstanding properties as labelling agents in long-term and deep tissue imaging due to their high chemical and photochemical stability, surpassing that of their oxygen- and silicon- substituted counterparts [1, 2, 3]. While recently many approaches to such NIR-absorbing fluorophores have been pursued, only a few examples are known in which these dyes were extended to fluorescent reporters, sensing and responding to specific physiological phenomena such as e.g. changes in the cell membranes potential [4, 5]. In this work we report the synthesis of the phosphorous rhodamine dye 1 for optical voltage sensing bringing together the advantages of POR dyes in terms of stability and NIR excitation with the functionality gained by the introduction of a lipophilic, conjugated molecular wire 3.
Voltage sensing dye 1 is synthesized over seven steps, of which three steps are the convergent synthesis of molecular wire 3. The final POR dye 1 is designed to intercalate into the cell membrane in a manner that the electron donating dimethylamine reaches into the cytoplasm enabling voltage sensing while the charged POR head of the molecule prevents cellular uptake.
References:
Grzybowski, M. et al.: Angew. Chem. Int. Ed. 2018, 57, 10137-10141. 2. Grimm, J. B. et al.: Nat. Methods 2020, 17, 815-821.
Ogasawara H. et al.: Chem. Commun. 2018, 54, 299-302.
Huang, Y.; Walker, A. S.; Miller, E. W.: J. Am. Chem. Soc. 2015, 137, 10767-10776.
Gonzalez, M. A. et al.: J. Am. Chem. Soc. 2021, 143, 2304-2314.
Dr. Anna Junker – 24
Münster University
"Development of PET Tracers for the Imaging of CD73 Expression in Breast and Pancreatic Cancer"
Here we present the development of novel positron emission tomography (PET) tracers for the imaging of CD73 expression in triple-negative breast and pancreatic cancer and compare their performance with the PET imaging of FDG distribution in a xenograft breast cancer mouse model.
Development of PET Tracers for the Imaging of CD73 Expression in Breast and Pancreatic Cancer
A. Junker Münster/DE, C. Dobelmann Münster/DE, Georg Rolshofen Bonn/DE, Mirko Scortichini Bethesda/USA, K.A. Jacobson Bethesda/USA, Sonja Schelhaas Münster/DE, Sven Hermann Münster/DE, C.E. Müller Bonn/DE.
Dr. Anna Junker, University of Münster, European Institute for Molecular Imaging, Waldeyerstr. 15, 48149 Münster
The 5’-ectonucleotidase (ecto-5’-NT, CD73) converts AMP to immunosuppressive adenosine. Numerous studies have demonstrated the overexpression of CD73 in the tumor microenvironment (TME) and its crucial role in cancer cell proliferation, tumor angiogenesis, and tumor immune escape processes [1]. Therefore, CD73 has become an appealing prognostic marker and therapeutic target.
Here we present the development of novel positron emission tomography (PET) tracers 1 and 2 for the imaging of CD73 expression in triple-negative breast and pancreatic cancer and compare their performance with the PET imaging of FDG distribution in a xenograft breast cancer mouse model [2].
Fig 1. The novel CD73- targeting PET tracers 1 and 2. In vivo PET imaging in tumor- bearing mice (s.c. MDA-MB-231 xenografts, left and right shoulder). Representative PET images 4h p.i. demonstrate a pronounced accumulation of the tracer in the tumor xenografts (white arrows) that was diminished in a blocking study.
References:
H. Z. Zhang, H. Z., Biomed. Res. Int. 2014, 460654.
Junker, Anna, et al. EP 22 157 311.6.
About the Author
Dr. Anna Junker is an Emmy Noether research group leader at the European Institute for Molecular Imaging at Muenster University.
"Exploring nucleic acid stability in Ionic Liquids: Measurements and mechanisms"
Exploring nucleic acid stability in Ionic Liquids: Measurements and mechanisms
Rajani K, Sanjib Senapati1
Department of Biotechnology, Indian Institute of Technology, Madras India
The stability and integrity of nucleic acids are vital in biotechnological applications. Conventionally, nucleic acids are stored under refrigeration as they are not stable at room temperature. Repeated freeze- thaw cycles are detrimental to the stability of nucleic acids and remain a cost ineffective way of preservation. Ionic liquids, comprised of organic cations and organic/inorganic anions are promising solvent media for preservation of biomolecules. Previously, we have shown that groove binding of IL cations contribute significantly to the DNA stability. [1] In this work, we attempt to understand the role of different class of ILs in stabilizing DNA through experimental methods and explore their mechanism through Molecular Dynamic simulations. The ILs of different class that are chosen involve protic, aprotic and borderline ILs. We employed UV-Visible absorption spectroscopy and CD spectroscopy to investigate the interaction pattern between DNA and ILs and measure the structural integrity. The melting values (Tm) were recorded as a measure of stability. Further, we performed MD simulations and quantum calculations to understand the mechanism of interaction of ILs with DNA. Our findings show the significance of protic IL, [EAN] in stabilizing DNA to a greater extent with highest Tm value and a free energy of stabilization.
Reference:
Chandran A., Ghoshdastidar D., Senapati S. Groove binding mechanism of ionic liquids: A key factor in long-term stability of DNA in hydrated ionic liquids? J. Am. Chem. Soc. 2012,134(50), 20330-20339
Özlem Kalkan – 25
European XFEL
"Overexpression, purification and structural analysis of a functional slime mold dye-decolorizing peroxidase with an improved heme stoichiometry"
Overexpression, purification and structural analysis of a functional slime mold dye-decolorizing peroxidase with an improved heme stoichiometry
Özlem Kalkan1,2, Sravya Kantamneni1, Adrian P. Mancuso1,3, Faisal H. M. Koua1
European X-ray Free Electron Laser GmbH, Holzkoppel 4, D-22869 Schenefeld, Schleswig-Holstein, Germany.
Istanbul University, Faculty of Science, Department of Molecular Biology and Genetics, 34134, Istanbul, Turkey. Email: ozlem.kalkan@xfel.eu
La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Victoria 3086, Australia.
Slime mold Dye-decolorizing peroxidase (smDyPA) is a newly discovered heme peroxidase, which catalyzes the H2O2-mediated oxidation of a wide-range of substrates such as polycyclic dyes and lignins, holding promises for potential industrial applications. To study the molecular mechanism of smDyPA, a highly pure and functional heme-bound protein is required, however, obtaining the correct heme stoicheometry is challenging and often requires a heme- reconstitution during purification. Here we show that a heme-bound peroxidase is temperature and medium dependent. Overexpressing smDyPA in enriched medium at low-temperatures is essential for obtaining functional heme-bound smDyPA. UV-visible spectroscopy on purified smDyPA showed a strong soret peak at 400 nm and a Reinheitszahl (Rz) (AbsSoret/Asb280) ratios of 0.75–1.0 when expressed in an enriched medium at 20°C in comparison with ~0.2 Rz value at high-temperatures. Furthermore, a crystallization of functional smDyPA yielded a brwonish crystal indicating a heme-binding. Crystal with 105 × 45 × 15 μm3 size diffracting X- rays to ~1.85 Å resolution was obtained, which is belong to the primitive tetragonal P43 21 2 spacegroup. The crystal structure reveals a heme molecule with full occupancy; in addition to the presence of a dioxygen with O–O distance of 1.27 Å indicating a formation of heme Fe(III)- Superoxo complex.
Aidan Kerckhoffs
"Red-shifting azobenzenes: periodic trends, tuneable half-lives and chalcogen bonding"
Red-shifting azobenzenes: periodic trends, tuneable half-lives and chalcogen bonding
Kerckhoffs,1 K. Kirstensen1 and M. Langton1,2
Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK
Azobenzenes are robust photoswitches operating with UV light, which damages cells and exhibits poor tissue penetration. [1] ABs operating within the red to near-IR region with controllable thermal relaxation rates are thus attractive components for photo-regulating biological processes. Ortho substituents destabilise the n molecular orbital of ABs; allowing switching in the visible region by exciting the n → π* transition. [2]
Herein, we report the first examples of heavy chalcogen and halogen tetra-ortho azobenzenes. All derivatives feature functional handles for further functionalisation. We report robust periodic trends for the chalcogen and halogen tetra ortho-substituted azobenzene series, where heavier atoms can be used to the tune switching wavelengths to the red and near-IR range due to increased steric clash with the diazo group. For the o-chalcogens, intramolecular chalcogen bonds lead to higher populations of planar (and thus more red-shifted) conformations; as confirmed in the solid-state. Remarkably, the rate of the Z → E thermal isomerisation can be tuned over seven orders of magnitude across the series.
We anticipate these tuneable, robust and biocompatible photo-switches will find utility within photo- pharmacology and other applications in which the precise tuning of photo-switching and thermal relaxation within otherwise isostructural molecular switches is desired.
References:
R, V. Jerca, F. Jerca, Nat. Rev. Chem. 2022, 6, 51-69
Konrad, D. Trauner et al, J. Am. Chem. Soc. 2020, 142, 14, 6538-6547
Moritz K. T. Klischan – 26
Heinrich Heine University Düsseldorf
"8,8’-Biflavones: Bioactivity Comparison of Flavone Dimers with Monomers"
A library of natural biflavone analogues was synthesized in a scalable manner. The bioactivity against different targets was assessed and the dimers compared with the monomers.
8,8’-Biflavones: Bioactivity Comparison of Flavone Dimers with Monomers
Moritz Klischan1, Flaminia Mazzone2, Julian Greb1, Lena Berning3, Max Schlamkow1,4, Björn Stork3, Klaus Pfeffer2, Jörg Pietruszka1,4*
Institute of Bioorganic Chemistry, Heinrich-Heine University Düsseldorf, Forschungszentrum Jülich, Stetternicher Forst, Geb.15.8, 52426 Jülich (Germany);
Institute of Medical Microbiology and Hospital Hygiene, Heinrich-Heine-University Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf (Germany);
Institute of Molecular Medicine I, Heinrich-Heine-University Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf (Germany);
Institut für Bio- und Geowissenschaften (IBG-1: Bioorganische Chemie) Forschungszentrum, 52428 Jülich (Germany);
Email of corresponding author: m.klischan@fz- juelich.de;
8,8’-Biflavones are an elusive class of flavonoid natural products first isolated in 1966.[1] As of today, there have been several procedures for the synthesis of the naturally occurring Cupressuflavone (CUF). To the best of our knowledge only one attempt has been made to synthesize a non-natural derivative.[2] Activities against Toxoplasma gondii proliferation were investigated and the selectivity indices of the flavone dimers compared with their monomer counterparts. A key step is the regioselective oxidative coupling of the acetophenones to form the acetophenone dimers.[3] These key-intermediates are a reoccurring motif in different polyketide-based natural products.[4] DFT-calculations have been conducted to rationalize the regioselectivity of this coupling.[5] We present the robust, scalable synthesis of a diverse library of bioactive biflavones. We show that scalability of the reactions and diversification and untapped potential of this compound class may result in further investigations of their bioactivities.
References:
V. Murti, P. Raman, T. J. Seshadri, Tetrahedron 1967, 23, 397-404.
H. Kikuchi, T. Hoshikawa, S. Kurata, Y. Katou, Y. Oshima, J. Nat. Prod. 2016, 79, 1259-1266.
K. Tanaka, F. Toda, Molecular Crystals and Liquid Crystals 1990, 187, 49-52.
For an example see: Wu, T. Iwata, A. Scharf, T. Qin, K. D. Reichl, J. A. Porco Jr, J. Am. Chem. Soc. 2018, 140, 5969-5975.
M. Liljenberg, J. H. Stenlid, T. Brinck, The Journal of Physical Chemistry A 2018, 122, 3270- 3279.
About the Authors
The authors are part of the GRK2158 “Natural products and natural product analogs against therapy-resistant tumors and microorganisms: new lead structures and modes of action” an interdisciplinary cluster located at the Heinrich-Heine University.
Amsterdam UMC, VU University, Department of Radiology and Nuclear Medicine
"Synthesis, in vitro and in vivo evaluation of novel radioligands for α-synuclein PET imaging"
Presently, the accurate clinical diagnosis of α-synucleinopathies is limited to histological examination of postmortem human tissue. Molecular imaging of α-synuclein in living human brains by positron emission tomography (PET) is not yet possible because a suitable tracer is still missing. The aim of the present study is to develop an α-synuclein small molecule PET tracer.
Synthesis, in vitro and in vivo evaluation of novel radioligands for α-synuclein PET imaging
Špela Korat1,2, Pedro M. Pereira1,2, Ran Sing Saw3, Kristina Herfert3, Wissam Beaino1,2, Benny Bang-Andersen4, Iwan J. P. de Esch5, Danielle J. Vugts1,2, Albert D. Windhorst1,2
Amsterdam UMC location Vrije Universiteit Amsterdam, dept Radiology & Nuclear Medicine, De Boelelaan 1117, 1081HV Amsterdam, The Netherlands;
Amsterdam Neuroscience, Brain imaging, Amsterdam, The Netherlands
Werner Siemens Imaging Center, Department of Preclinical Imaging and Radiopharmacy, Eberhard Karls University Tübingen, Tübingen, Germany
H. Lundbeck A/S, Valby, Denmark
Division of Medicinal Chemistry, Amsterdam Institute for Molecules, Medicines and Systems, VU Amsterdam, De Boelelaan 1108, 1081HV Amsterdam, The Netherlands
Aggregates of α-synuclein are a hallmark of several neurodegenerative diseases, including Parkinson’s disease.1 Non-invasive quantitative assessment of α-synuclein in vivo using positron emission tomography (PET) could provide valuable insights into the early diagnosis, however a suitable PET tracer does not exist.2 We applied in silico design using a pharmacophore model based on published α-synuclein small molecule ligands to define new leads. Flexible alignment with pharmacophoric consensus using MOE 2018.01 was performed to achieve a pharmacophore model that identifies key steric and electronic features. Based on these results, a series of compounds was proposed, synthesized and evaluated using α-synuclein recombinant fibrils. The most potent ligands (Ki < 20 nM) were radiolabeled with tritium and evaluated with in vitro autoradiography (using brain tissue from a deposition mouse model and postmortem human brain). Three compounds showed specific binding to α-synuclein and were radiolabeled with carbon-11. Brain uptake of the 11C-labelled compounds was evaluated in vivo in wild-type C57Bl6/J mice and showed sufficient brain uptake. Further in vivo evaluation, such as ex vivo biodistribution and metabolic stability, is currently underway.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 813528.
References:
Spillantini,M. et al.Nature 1997, 388, 839–840;
Korat,Š. et al.Pharmaceuticals 2021, 14, 847;
About the Author
Špela Korat is a PhD candidate at the Amsterdam UMC, Department of Radiology and Nuclear Medicine. Currently, she is working on a project that focuses on PET tracer development for α-synuclein.
Faisal H. M. Koua – 29
European XFEL GmbH
"Redox structures of high potential iron sulfur protein revealed by megahertz serial femtosecond crystallography"
We resolved the redox structures of High-potential iron sulfur protein using room-temperature serial femtosecond crystallography with ultra-short (~20 fs) X-ray pulse duration resulting in radiation-damage free models.
Redox structures of high potential iron sulfur protein revealed by megahertz serial femtosecond crystallography
F.H.M. Koua1, I.E. Dawod1,2, J. Bielecki1, M. Kloos1, E. De Santis2, D. Melo1, N.-E. Christou3, K. Dörner1, A. Sarma1, M. Sikorsky1, R. Letrun1, M. Vakili1, S. Kantamneni1, J.C.P. Koliyadu1, C. Kim1, P. Vagovic1,3, T. Sato1, A. Round1, H. Han1, R.J. Bean1, J. Schulz1, A.P. Mancuso1,4
European XFEL GmbH, Schenefeld, Schleswig-Holstein, Germany;
Department of Physics and Astronomy, Uppsala University, Uppsala, Sweden;
Center for Free-Electron Laser Science, Deutsches Elektronen-Synchrotron, Hamburg, Germany;
La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Australia. Email: faisal.koua@xfel.eu
Keywords: Serial femtosecond crystallography, Metalloproteins, Iron-sulfur clusters, Radiation-damage
The high-potential iron sulfur protein (HiPIP), an electron transfer mediator in photosynthetic bacteria, is a small soluble metalloprotein harboring a single iron-sulfur (Fe4S4) cluster in its molecular center. HiPIP has a high redox potential (E0 ≥ +350 mV) comparing to other protein-containing Fe4S4 clusters such as ferredoxin which has an E0 ≈ –400 mV [1,2]. This high redox potential is achieved by the transition between the resting (reduced) [Fe4S4]2+ and active (oxidized) [Fe4S4]3+ states which is governed by the hydrogen-bonding network within the local vicinity of the cluster [3]. However, resolving such atomic details with the correct geometry of the metal cluster are often hampered by X-ray radiation damage [4]. Here, we report the first radiation-damage free structures of the redox states of HiPIP at ~1.65 Å resolution using room-temperature serial femtosecond crystallography (SFX) with X-ray pulses shorter than 25 fs at megahertz intra-train repetition rates. The results reveal that most of the Fe–S/Fe–Cysteine(S) intra-atomic distances are shortened by 0.03–0.1 Å in comparison with the synchrotron models indicating that the SFX models are less affected by X-ray radiations. Our SFX models will provide an exemplar for metalloproteins enabling further theoretical and experimental studies on redox enzymology.
References:
Hirano, Y., Takeda, K. & Miki, K. (2016). Nature 534, 281–284.
Beinert, H. (2000). Biol. Inorg. Chem. 5, 2–15.
Carter, C.W. Jr. (2001). Handbook of metalloproteins, edited by A. Messerschmidt, R. Huber, T.L. Poulos & K. Weighardt, pp. 602–609. New York:
Yano, J., Kern, J., Irrgang, K.D., Latimer, M.J., Bergmann, U., Glatzel, P., Pushkar, Y., Biesiadka, J., Loll, B., Sauer, K., Messinger, J. Zouni, A. & Yachandra, V.K. (2005). Proc. Natl. Acad. Sci. USA,102,
Removal of aberrant aggregates in amyloid diseases: from in silico analysis to in vitro biological activity
In silico identification of physicochemical and structural rules that govern the ability of certain compounds to disaggregate preformed amyloid fibrils. The most promising pre-selected compounds are currently under evaluation in experimental assays against preformed amyloid fibrils of transthyretin, as well in some other amyloid precursors.
Removal of aberrant aggregates in amyloid diseases: from in silico analysis to in vitro biological activity
Zaida L. Almeida and Rui M. M. Brito
Chemistry Department and Coimbra Chemistry Centre-Institute of Molecular Sciences (CQC-IMS), University of Coimbra, 3004-535 Coimbra, Portugal
Amyloidoses are highly debilitating and incurable diseases related with formation and deposition of insoluble amyloid fibrils1. Today, approximately 60 amyloidosis are known associated with more than 40 different proteins.
One of the therapeutic strategies being explored to overcome amyloid deposition is amyloid disaggregation2. However, and despite the recent progresses in understanding amyloidosis, most therapeutic efforts related to amyloid disaggregation have failed.
In this work, we used in silico methods to characterize the physicochemical determinants and structural rules that govern the ability of certain molecules to disaggregate amyloid. Firstly, we performed an extensive literature search to identify compounds with amyloid fibril disruption activity. 606 compounds with activity against 29 amyloid precursor proteins were selected. This curated chemical library was then enriched with compound information such as: physicochemical properties, toxicity, and drug-like profile. Compounds were also clustered based on structural similarity.
Independently, an in vitro amyloid disaggregation screening protocol was developed3 to test the activity of compounds prioritized from the computational analysis. Several compounds were tested in early and late stages of amyloid formation by transthyretin, a well-known protein involved in several amyloidosis. DLS and TEM were also used to characterize the effect of the selected compounds on TTR amyloid disaggregation.
Ferreira, E.; Almeida, Z.L.; et al. Int. J. Mol. Sci.2022, 23(1), 391.
About the Author
Zaida L. Almeida is currently finishing her PhD in the field of Biological Chemistry. Her research interests are focused on protein misfolding and amyloid fibril formation, as well as on drug design against amyloid-based diseases.
"Re-use of Caco-2 monolayers in permeability assays – validation regarding cell monolayer integrity, biochemistry, and function"
Developing of an experimental procedure that allows the use of Caco-2 monolayers for three independent permeability assays during the period of 21 to 30 days after cell seeding. The validation of the re-use procedure was performed regarding the Caco-2 monolayers integrity, biochemistry, and function.
Re-use of Caco-2 monolayers in permeability assays – validation regarding cell monolayer integrity, biochemistry, and function
Cristiana L. Pires,1 Ana L. M. Batista de Carvalho,2 Lino Ferreira,3 Maria Paula M. Marques, 2,4 and Maria João Moreno1
CQC-IMS, Coimbra Chemistry Centre – Institute of Molecular Sciences, University of Coimbra, 3004-535 Coimbra, Portugal
Molecular Physical-Chemistry R&D Unit, Department of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal
CNC-Center for Neuroscience and Cell Biology, CIBB-Centre for Innovative Biomedicine and Biotechnology, University of Coimbra, Biotech Parque Tecnológico de Cantanhede, 3060-197 Coimbra, Portugal
Department of Life Sciences, University of Coimbra, 3000-456 Coimbra, Portugal
Caco-2 monolayers are commonly used to predict the intestinal drug absorption in humans. The reference protocol1 requires 21 days to establish a fully differentiated and confluent cell monolayer, which is used in a single permeability assay during the period of monolayer stability (21-30 days)2. To increase the throughput of this assay, we developed a protocol that allow the use of cell monolayers for three permeability assays. The procedure includes the incubation of the cell monolayer in culture medium for two full days after the permeability assays, with assays being performed on days 22, 25 and 28. The integrity of the cell monolayer have been guaranteed by the low Papp values of Lucifer yellow obtained for single use and re- used cell monolayers3.
In this work, we performed a robust validation of the re-use procedure regarding the transcellular transport. The permeability of several model drugs following distinct permeation routes were carried out both in single use and re-used cell monolayers. Additional validation regarding the functional properties was provided with the evaluation of the expression and activity of the efflux transporter P-glycoprotein. The full validation of the re-use of Caco- 2 monolayers demonstrates the utility of this new procedure to triplicate the throughput of the assay.
References:
Hubatsch, I., Ragnarsson, E. G. E., Artursson, P. Nature Protocols, 2007, 2, 2111-2119
BriskeAnderson, M. J., Finley, J. W., Newman, S. M. Proceedings of the Society for Experimental Biology and Medicine1997, 214, 248-257
Pires, C. L.; Praça, C.; Martins, P. A. T.; Batista de Carvalho, A. L. M.; Ferreira, L.; Marques, M. P. M.; Moreno, M. J. Pharmaceutics2021, 13, 1563
About the Author
Cristiana L. Pires is currently finishing her PhD degree in Biological Chemistry. She focused on improve the use of in vitro permeability data in the generation of in silico models to predict the permeability of new drugs across biological barriers.
Acknowledgments
This research was supported by the Portuguese “Fundação para a Ciência e a Tecnologia” (FCT) through projects UIDB/00070/2020, UIDB/00313/2020, PT2020-PTDC/QUI-OUT/29373/2017, PT2020-PTDC/DTP-FTO/2784/2014 and fellowship SFRH/BD/138873/2018.
Johannes Lang – 32
University of Heidelberg
"Diversification of the Phenylglycine Scaffold in Dengue Virus Protease Inhibitors"
Diversification of the Phenylglycine Scaffold in Dengue Virus Protease Inhibitors
Johannes Lang, Lisa Reichert, Nikos Kühl, Christian D. Klein*
Medicinal Chemistry, Institute of Pharmacy and Molecular Biotechnology IPMB, Heidelberg University, Im Neuenheimer Feld 364, D-69120 Heidelberg, Germany
Dengue virus is one of the most challenging threats to global health, and antiviral drugs with activity against dengue or related flaviviruses are urgently required. Viral enzymes such as the NS2B-NS3 protease are promising targets for the development of specific antiviral compounds.1 We recently published phenylglycine derivatives2,3 which had considerably higher activity in a cell-based dengue protease reporter gene assay (DENV2proHeLa) than earlier covalent and non-covalent peptidic inhibitors.4,5
In this work, further structure-activity relationship studies on the phenylglycine’s amine cap were conducted. 60 different moieties were introduced using solid phase peptide synthesis. While bulky aromatic groups are preferred in this position, sterically hindered amides are also accepted. However, physicochemical properties such as lipophilicity could not be sufficiently addressed with this structural exploration.
To overcome this challenge, the phenylglycine core was replaced by around 25 natural and non-natural aliphatic and aromatic amino acids. Lipophilic aromatic and aliphatic residues are accepted in this position and lead to single-digit micromolar EC50 values in the cellular DENV2proHeLa assay. Moieties with a moderately increased polarity such as tryptophan or a phenylated histidine were also tolerated. These structural elements could serve as basis for new classes of NS2B-NS3 protease inhibitors with increased drug-likeness.
References:
Behnam, M. A. M.; Nitsche, C.; Boldescu, V.; Klein, C. D., The Medicinal Chemistry of Dengue Virus. Journal of Medicinal Chemistry 2016, 59 (12), 5622-5649.
Kühl, N.; Graf, D.; Bock, J.; Behnam, M. A. M.; Leuthold, M.-M.; Klein, C. D., A New Class of Dengue and West Nile Virus Protease Inhibitors with Submicromolar Activity in Reporter Gene DENV-2 Protease and Viral Replication Assays. Journal of Medicinal Chemistry 2020, 63 (15), 8179-8197.
Kühl, N.; Leuthold, M. M.; Behnam, M. A. M.; Klein, C. D., Beyond Basicity: Discovery of Nonbasic DENV-2 Protease Inhibitors with Potent Activity in Cell Culture. Journal of Medicinal Chemistry 2021, 64 (8), 4567-4587.
Behnam, M. A. M.; Graf, D.; Bartenschlager, R.; Zlotos, D. P.; Klein, C. D., Discovery of Nanomolar Dengue and West Nile Virus Protease Inhibitors Containing a 4- Benzyloxyphenylglycine Residue. Journal of Medicinal Chemistry 2015, 58 (23), 9354-9370.
Nitsche, C.; Zhang, L.; Weigel, L. F.; Schilz, J.; Graf, D.; Bartenschlager, R.; Hilgenfeld, R.; Klein, C. D., Peptide–Boronic Acid Inhibitors of Flaviviral Proteases: Medicinal Chemistry and Structural Biology. Journal of Medicinal Chemistry 2017, 60 (1), 511-516.
Stefan Laufer – 33
Tuebingen University
"Tübingen Center for Academic Drug Discovery TüCAD2 - Drug Innovation in Academia"
TüCAD2 is an Academic Drug Discovery Platform, based on pharmacological validation of genetically identified targets. Up to now, four compounds of this platform made it first into man.
Tübingen Center for Academic Drug Discovery TüCAD2
Lars Zender1,3 , Stefan Laufer2,3,4
Department of Medical Oncology and Pneumology, University Hospital Tuebingen, Tuebingen, Germany
Medicinal Chemistry, Dept. of Pharmacy & Biochemistry, Eberhard Karls University Tübingen, Auf der Morgenstelle 8, 72076 Tübingen,
IFIT Cluster of Excellence EXC 2180 ‘Image-Guided and Functionally Instructed Tumor Therapies’, University of Tuebingen, Tuebingen, Germany.
Tübingen Center for Academic Drug Discovery & Development (TüCAD2), Tübingen, Germany
Our research in RNAi- and Crispr/Cas9-based functional genomics especially focuses on the identification of new cancer genes and therapeutic targets in therapy-resistant solid tumors. For such studies, clinically relevant mouse tumor models, which closely resemble the human disease, were are available. Specifically, we are combining so called mosaic mouse models with stable RNAi technology to dissect tumor suppressor networks in gastrointestinal tumors and to identify and validate new therapeutic target genes. Together with a limited number of other laboratories worldwide, we have the expertise to conduct RNAi screens for new cancer genes directly in orthotopic and immunocompetent cancer mouse models in vivo.
To best translate data from our unique RNAi platform into new cancer therapies, we recently systematically connected our RNAi expertise with the research areas virtual screening/modelling and medicinal chemistry to build an academic drug discovery unit, designated TüCAD2 (Tübingen Centre for Academic Drug Discovery). Our unit was recently approved as a member of the worldwide acting Academic Drug Discovery Consortium (AD2C, http://addconsortium.org/drug-discovery-factsheet.php?ddc_id=DC1000196). TüCAD2 represents an interfaculty and interdisciplinary endeavor and was founded by the Dept. of Pharmaceutical/Medicinal Chemistry (Stefan Laufer) and the Dept. of Internal Medicine VIII (Lars Zender).
We will discuss the pivotal role of academic drug discovery infrastructures for rapidly translating validated therapeutic target structures into clinical applications and will give an example of a novel and promising drug for the treatment of liver cancer which entered the phase of clinical testing only 13 months after completion of pivotal preclinical proof of concept.
Founded in 20212 and being part of Tübingen’s Excellence Strategy, TüCAD2 has been able to bring four compounds first into man and has supported two VC-financed biotech startups
About the Author
Stefan Laufer is Professor and Chairman for Pharm./Med. Chemistry at Tuebingen University, Director of TüCAD2. He is associate editor of ACS J. Med. Chem with >600 publications.
His work is highly translational and yielded 5 cpds. first into man.
Emilio Lence – 34
Universidade de Santiago de Compostela
"Simulation Studies with Bicyclic Boronate β-Lactamase Inhibitors to Identify the Molecular Basis of Its Ultrabroad Efficacy"
Simulation Studies with Bicyclic Boronate β-Lactamase Inhibitors to Identify the Molecular Basis of Its Ultrabroad Efficacy
Emilio Lence,1 and Concepción González-Bello1
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela. Jenaro de la Fuente s/n, Santiago de Compostela 15782, Spain e-mail: emiliojose.lence@usc.es
The clinical utility of β-lactam antibiotics, which represent about 70% of all antibacterial drugs, is currently threatened by the worldwide proliferation and dissemination of bacteria producing β-lactamases.1 These enzymes that hydrolysed their β-lactam ring, is the most relevant resistance mechanism in deadly pathogens such as Pseudomonas aeruginosa, Acinetobacter baumannii and Enterobacteriaceae. β-lactamases presents huge structural diversity and hydrolytic capability; with enzymatic mechanisms, involving covalent (serine-β-lactamases) or non-covalent catalysis (metallo-β-lactamases). In top of this, the emergence of multi-drug resistance bacteria capable of simultaneously producing diverse β-lactamases, makes the development of an efficient ultrabroad-spectrum β-lactamase inhibitor a challenging task. A recent breakthrough in the field was the discovery of bicyclic boronate inhibitors, such as taniborbactam (TANI) and xeruborbactam (XERU). They mimic both transition state analogs in serine- and metallo-β-lactamases. Between both, only XERU is clinically useful against bacterial strains producing class D carbapenemases. The outstanding inhibitory properties of XERU against metallo-β-lactamases (IMP-1) and the most challenging class D carbapenemases (OXA-48, OXA-23) attracted our attention. To understand the molecular basis of the activity of XERU, we have studied the formation of the corresponding enzyme-boronate adducts/complexes with these targets and we have compared with TANI and other cyclic boronates by Molecular Dynamic simulation.2,3
Financial support from the Spanish Ministry of Science and Innovation (PID2019-105512RB-I00/AEI/10.13039/501100011033), the Xunta de Galicia [ED431C 2021/29 and Centro singular de investigación de Galicia accreditation 2019–2022 (ED431G 2019/03)], and the European Regional Development Fund is gratefully acknowledged.
References
C. González-Bello, et al. J. Med. Chem.2020, 63, 1859-1881.
E. Lence, C. González‐Bello Adv. Therap.2021, 2000246
E. Lence, C. González‐Bello. Front. Microbiol.2021, 12, 721826.
Elina Lidumniece – 35
Latvian Institute of Organic Synthesis
"Rationally designed inhibitors of malarial subtilisin-like serine protease containing boronic acid warhead"
In this poster we present the synthesis and inhibitory activity of peptidic boronic acids against malarial subtilisin-like serine protease (SUB1).
Rationally designed inhibitors of malarial subtilisin-like serine protease containing boronic acid warhead
Elina Lidumniece1, Chrislaine Withers-Martinez2, Michael J. Blackman2, Aigars Jirgensons1
Latvian Institute of Organic Synthesis, Riga LV-1006, Latvia
Malaria Biochemistry Laboratory, The Francis Crick Institute, London NW1 1AT, United Kingdom
All clinical manifestations of malaria are caused by cycles of parasite proliferation within red blood cells. Subtilisin-like serine protease (SUB1) is a multifunctional processing protease which palys a significant role in merozoite egress (Figure 1) by activation of a cascade of proteolytic events, leading to rupture of human red blood cell (RBC). Inhibition of SUB1 can prevent from parasite replication and disease progression rendering this enzyme as an attractive drug target1.
Figure 1. The merozoite egress from red blood cell.
Previous research revealed boronic acid based inhibitor 1 with low nanomolar PfSUB1 inhibitory potency and remarkable inhibition of parasite egress in RBC assay2 (Scheme 1). Inhibitor 1 was selected as a base for further development of SUB1 inhibitors by modifying P1, P3 and P5 positions. Here we present the inhibitory activity of synthesized compounds compared to the parent inhibitor 1.
Scheme 1. Development of peptidic boronic acid containing inhibitors.
Lidumniece, E.; Withers-Martinez, C.; Hackett, F.; Collins, C. R.; Perrin, A. J.; Koussis, K.; Bisson, C.; Blackman, M. J.; Jirgensons, A. Natl. Acad. Sci. U. S. A., 2021, 118, e2022696118
About the Author
Elina Lidumniece is a Ph.D. student at Riga Technical University and works at the Latvian Institute of Organic Synthesis with the guidance of Prof. Aigars Jirgensons on the synthesis and design of new malarial enzyme PfSUB1 inhibitors.
Department of Biosciences and Bioengineering, Indian Institute of Technology Roorkee, Roorkee, Uttarakhand, 247667, India
ICAR- Central Citrus Research Institute, Nagpur, Maharashtra, 440010, India
Email: slonare@bt.iitr.ac.in
Citrus Huanglongbing (HLB) disease is caused by Candidatus Liberibacter asiaticus (CLas), an unculturable phloem-restricted Gram negative bacterium which is transmitted by the Asian citrus psyllid (ACP), Diaphorina citri. CLas is depends upon ACP to move in plant hosts and perhaps this contributes to its long-term survivability. Till date no effective control strategies have been developed for management of HLB disease. Effective strategies to control CLas could include the development of inhibitor molecules/antimicrobials against the CLas proteins critical in bacterial survival through proteome and in-silico analysis. ATP-Binding Cassette (ABC) transporters which are integral membrane proteins mainly facilitates the transfer of diverse substrates across the membrane using energy provided by ATP hydrolysis. In this work, one of such CLas ABC transporter protein which is cystine binding protein was cloned, expressed and purified using three steps purification and also characterization of such CLas protein was carried out. The bioinformatics based virtual screening was employed to screen the potential inhibitor molecules. We have also determined the crystal structure of CLas protein in complex with inhibitors with a resolution of nearly 2.50 Å. Such inhibitors molecules are evaluated by various biophysical techniques. The evaluation of selected lead molecules is carried out, in planta, under controlled conditions to get rid of HLB disease as ABC transporter proteins are potential drug target.
Fernanda Manaia – 12
São Paulo State University
"Benzofuroxan analogues are highly active and selective against Mycobacterium tuberculosis"
Benzofuroxan analogues are highly active and selective against Mycobacterium tuberculosis.
Fernanda Manaia Demarqui1, Guilherme Felipe dos Santos Fernandes1, Vitória Yumi Kinoshita1, Jean Leandro dos Santos1, Fernando Rogério Pavan1
São Paulo State University (UNESP), School of Pharmaceutical Sciences, Araraquara 14800903, Brazil
The established treatment for tuberculosis disease has been used for more than 50 years and lasts 6 months for susceptible strains of Mycobaterium tuberculosis but can reach over 2 years in cases of resistance, with the introduction of new classes of antibiotics. In recent years, efforts to find new treatments for tuberculosis have increased. Recently described benzofuroxan compounds have in vitro and in vivo activity, presenting sterilizing antitubercular activity. The compounds BF07, BF 08, BF09, BF10 and BF14, analogous to these, had their in vitro activity against M. tuberculosis and their cytotoxicity in mammalian cells evaluated. The resazurin microtiter assay was performed with the H37Rv strain to evaluate in vitro activity. For the cytotoxicity assay, MRC-5 and J774.A cell lines were used. The Minimum Inhibitory Concentration of the compounds was 0.4 μg/ml for BF08, 0.33 μg/ml for BF09 and 4.75 μg/ml for BF14, no activity was observed for BF07 and 10. In the lung fibroblast and macrophage lineages tested, the compounds were not cytotoxic in a concentration of 100 μg/ml and, therefore, presented a high selectivity index. These results show that benzofuxorans and their analogues are a promising path in the search for new antibiotics against tuberculosis.
María Maneiro Rey – 36
Universidade de Santiago de Compostela
"Target-activation of Cyclic sp3-hybridized Carbons for Irreversible Inhibition of an Anti-virulence Target"
Target-activation of Cyclic sp3-hybridized Carbons for Irreversible Inhibition of an Anti-virulence Target
María Maneiro,1 Ángela Rodríguez,1 Emilio Lence,1 José Manuel Otero,1 Mark J. van Raaij,2 and Concepción González-Bello1,*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, Santiago de Compostela, Spain. E-mail: concepcion.gonzalez.bello@usc.es
Departamento de Estructura de Macromoléculas, Centro Nacional de Biotecnología (CSIC), Madrid, Spain.
The development of selective irreversible inhibitors is a challenge since the introduction of overly reactive electrophiles in the scaffold might increase the occurrence of undesired off-target effects. A booming strategy involves the use of barely reactive electrophilic groups that are only activated towards reaction upon binding to their target. The correct arrangement of the ligand and suitable geometry of the electrophilic group relative to the enzyme/protein nucleophile are key for successful covalent modification. Targeting ligand carbon atoms with little electrophilic character (sp3), appears a challenging task that although little explored, might expand our options for drug design. Herein we study the activation of cyclic sp3-hybridized carbons for irreversible inhibition of a promising target for anti-virulence drug development, the type I dehydroquinase (DHQ1) enzyme.1 Compounds 1–2, bearing an amino functionality into the quinate scaffold, seek the enhancement of the electrophilicity of C3 towards covalent modification.2 The reactivity of C3 was also explored by introduction of an oxirane moiety with an S,S configuration at the C2-C3 bond (compound 3) to compare the geometry requirements of the C3 towards nucleophilic attack. A combination of structural, biochemical, and molecular dynamics simulation studies explained the molecular basis of the type of inhibition observed.
References:
(a) González-Bello, C. Future Med. Chem.2015, 7, 2371-2383. (b) González-Bello, C. et al. J. Am. Chem. Soc.2015, 137, 9333.
Rodríguez, Á. et al. Front. Mol. Biosci.2023, 10, 1111598.
Acknowledgements
Financial support from the Spanish State Agency of Research (PID2019-105512RB-I00/AEI/10.13039/501100011033), the Xunta de Galicia (ED431C 2021/29 and Centro singular de investigación de Galicia accreditation 2019-2022 (ED431G 2019/03), and the European Regional Development Fund (ERDF) is gratefully acknowledged.
Vanya Mantareva
Institute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences
"Synergistic Antitumor and Photo-safety Effects of Zn(II) Phthalocyanines and Co(III) Corrin (Vit. B12)"
Synergistic Antitumor and Photo-safety Effects of Zn(II) Phthalocyanines and Co(III) Corrin (Vit. B12)
Vanya Mantareva1,*, Ivan Iliev2, Inna Sulikovska2, Ivan Angelov1 and Mahmut Dumus3
Institute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Bld. 9, 1113 Sofia, Bulgaria; mantareva@yahoo.com
Institute of Experimental Morphology, Pathology and Anthropology with Museum, Bulgarian Academy of Sciences, Bld. 25, 1113 Sofia, Bulgaria
Gebze Technical University, Department of Chemistry, Gebze, 41400 Kocaeli, Turkey
*Correspondence: mantareva@yahoo.com; Tel.: ++ 35 9 9606 181
Photodynamic therapy (PDT) has been highlighted as a prompt curative method with minimum side effects and exclusively effective towards the fast development of multidrug resistance. The study presents Zn(II) phthalocyanines (ZnPcs) with peripheral and non-peripheral position of the substitution groups and cobalamin complex popular as vitamin B12 towards breast cancer cells. It was assumed that ZnPcs photocytotoxicity can be improved by addition of second porphyrinoid such as cobalamin due to non-covalent electrostatic and π–π interface interaction between both macrocycles. Two breast cancer cell lines (MDA-MB-231 and MCF-7) and a surrounding non-tumorigenic MCF-10A cell line were studied. BALB 3T3 cells were tested showing the photo-safety of compounds and the selectivity of cytotoxicity tumor vs. normal cells. The huge difference was observed at red light (660-nm) irradiation. The minimal cytotoxicity was observed due to the full solar spectrum of exposure for all tested cell lines. The synergistic antitumor efficiency was observed by combined application of both ZnPcs and cobalamin. However, the efficacy appeared not only due to photosensitization. As general, the PDT and a second photoactive compounds could have considerable outcome in diminishment of drug toxicity and the development of drug resistance in tumor cells.
Keywords: Phthalocyanines; Cobalamin, Vitamin B12; Photodynamic therapy (PDT); Breast cancer cells; Epithelial and fibroblast cell lines
Acknowledgements
Thanks to the project KP-06-H28/11, Bulgarian National Science Fund
José L. Medina-Franco – 37
Universidad Nacional Autonoma de Mexico (UNAM)
"Epigenetic drug discovery with epi-informatics"
We discuss the recent advances in epi-informatics to chart the epigenetic relevant space and guide the development of targeted libraries.
Epigenetic drug discovery with epi-informatics
José L. Medina-Franco
DIFACQUIM research group, Department of Pharmacy, School of Chemistry, National Autonomous University of Mexico (UNAM), Mexico City 04510, Mexico
A broad range of computational approaches collectively called “epi-informatics” are increasingly used to advance epigenetic drug and probe discovery [1]. Herein, we discuss the recent advances in epi-informatics to chart the epigenetic relevant space and guide the development of targeted libraries. We also discuss the applications of computational approaches to guide the identification of small molecules active against one or more epigenetic targets. In particular, we will cover current trends of machine learning models generated based on sizeable public compound databases annotated with biological activity and implemented in a free webserver [2]. As a case study, we will present the identification of potent and dual inhibitors of DNA and histone methyltransferases. In addition to showing low micromolar enzymatic inhibition, the small molecules are also active in various cell lines [3]. The hit compounds were identified from synthetic screening libraries focused on epigenetic targets after an exhaustive analysis of the diversity and coverage of the chemical space. Computational approaches helped to rationalize the activity at the molecular level.
Key words: artificial intelligence; chemoinformatics; machine learning; data visualization.
References:
Medina-Franco, J. L. Epi-Informatics: Discovery and Development of Small Molecule Epigenetic Drugs and Probes; Academic Press: London, UK,
Sánchez-Cruz N, Medina-Franco JL Epigenetic Target Fishing with Accurate Machine Learning Models. Med. Chem. 2021, 64, 8208–8220.
Medina-Franco JL, López-López E, Martínez-Fernández LP 7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1. Molecules 2022, 27,
About the Author
Dr. Medina-Franco leads the DIFACQUIM research group at UNAM in Mexico. We focus on chemoinformatics, molecular modeling and artificial intelligence with applications on epigenetic targets and natural products. See more at www.difacquim.com
"Differential Biophysical Interaction of Rifabutin on the Mycobacterial Membrane Envelope at Different Growth Stages"
Differential Biophysical Interaction of Rifabutin on the Mycobacterial Membrane Envelope at Different Growth Stages
Anjana P. Menon1,2,3, Tzong-Hsien Lee2,3, Marie-Isabel Aguilar2,3*, Shobhna Kapoor1,2,4*
Department of Chemistry, Indian Institute of Technology Bombay, Mumbai 400076, India
IITB-Monash Academy, Indian Institute of Technology Bombay, Mumbai 400076, India
Department of Biochemistry & Molecular Biology, Monash University, Clayton, Vic 3800, Australia
Graduate School of Integrated Sciences for Life, Hiroshima University, Hiroshima, 739-8528, Japan
Mycobacterial species possess a dynamic cell envelope consisting of an array of unique lipids, which contributes to the differential interaction with various therapeutic agents. The variation in the bacterial lipidome in the early and late infection stages further contributes to the need for critical analysis of the bacterial ability to tolerate antibiotic treatments under these conditions. In this work, a combination of biophysical techniques was performed using model membranes reconstituted by the lipids extracted from respective layers of the mycobacterial cell envelope, to be employed as a scaffold to screen for potential drug candidates for different infection stages. The UV-visible spectroscopic studies revealed that the outer membrane impacts on reduced rifabutin partitioning during late infection, while the fluorescence quenching studies exhibited limited rifabutin distribution across the inner membrane bilayer, further reducing the drug-entry into the mycobacterium1. The above studies show that the passive diffusion of this antitubercular drug, rifabutin is altered with the structural organization of the lipids in each leaflet, resulting in controlled drug-entry and hence, drug resistance. Extending these studies to a complete-lipid scaffold of mycobacterium species could pilot the designing of effectual drugs for the therapeutic eradication of mycobacterial species at different growth stages2.
References:
Menon AP, Dong W, Lee TH, Aguilar MI, Duan M, Kapoor S. Mutually Exclusive Interactions of Rifabutin with Spatially Distinct Mycobacterial Cell Envelope Membrane Layers Offer Insights into Membrane-Centric Therapy of Infectious Diseases. ACS bio & med Chem Au. 2022 Mar 24;2(4):395-408.
Amaral L, Viveiros M. Thioridazine: A non-antibiotic drug highly effective, in combination with first line anti-tuberculosis drugs, against any form of antibiotic resistance of Mycobacterium tuberculosis due to its multi-mechanisms of action. Antibiotics. 2017 Jan 14;6(1):3.
Aurélien F. A. Moumbock – 38
Albert-Ludwigs-Universität Freiburg
"ePharmaLib: A Versatile Library of e-Pharmacophores to Address Small-Molecule (Poly-)Pharmacology"
ePharmaLib is a pharmacophore library use to predict putative target proteins of bioactive compounds. It contains 15,148 epharmacophores, modeled from the crystal structures of pharmaceutically relevant protein–ligand complexes of the focused screening Protein Data Bank (scPDB) dataset.
ePharmaLib: A Versatile Library of e-Pharmacophores to Address Small-Molecule (Poly-)Pharmacology
Aurélien F. A. Moumbock1,2, Jianyu Li1, Hoai T. T. Tran1,3, Rahel Hinkelmann2, Evelyn Lamy3, Henning J. Jessen2, and Stefan Günther1
Institute of Pharmaceutical Sciences, Faculty of Chemistry and Pharmacy, Albert-Ludwigs-Universität Freiburg, Germany
Institute of Organic Chemistry, Faculty of Chemistry and Pharmacy, Albert-Ludwigs-Universität Freiburg, Germany 3Molecular Preventive Medicine, University Medical Center and Faculty of Medicine, Albert-Ludwigs-Universität Freiburg, Germany
Bioactive compounds oftentimes bind to several target proteins, thereby exhibiting polypharmacology. Experimentally determining these interactions is however laborious, and structure-based virtual screening (SBVS) of bioactive compounds could expedite drug discovery by prioritizing hits for experimental validation. Here, we present ePharmaLib, a library of 15,148 e-pharmacophores modeled from solved structures of pharmaceutically relevant protein–ligand complexes of the screening Protein Data Bank (sc- PDB). ePharmaLib can be used for target fishing of phenotypic hits, side effect predictions, drug repurposing, and scaffold hopping. In retrospective SBVS with compounds of the Streptomyces natural products database (StreptomeDB), a good balance was obtained between computational efficiency and predictive accuracy. As a proof of concept, we carried out prospective SBVS in conjunction with a photometric assay, which inferred that the mechanism of action of neopterin (an endogenous immunomodulator) putatively stems from its inhibition (IC50 = 18 μM) of the human purine nucleoside phosphorylase. This ready-to-use library is freely available at http://www.pharmbioinf.uni-freiburg.de/epharmalib.
Bibliography:
Moumbock AFA, Li J, Tran HTT, Hinkelmann R, Lamy E, Jessen HJ, Günther S. ePharmaLib: A Versatile Library of e-Pharmacophores to Address Small-Molecule (Poly-)Pharmacology. Chem. Inf. Model. 2021, 61(7): 3659-3666. doi: https://doi.org/10.1021/acs.jcim.1c00135. PMID: 34236848.
Moumbock AFA, Gao M, Qaseem A, Li J, Kirchner PA, Ndingkokhar B, Bekono BD, Simoben CV, Babiaka SB, Malange YI, Sauter F, Zierep P, Ntie-Kang F, Günther S. StreptomeDB 3.0: an updated compendium of streptomycetes natural products. Nucleic Acids Res. 2021, 49(D1): D600-D604. doi: https://doi.org/10.1093/nar/gkaa868. PMID: 33051671
Moumbock AFA, Li J, Mishra P, Gao M, Günther S. Current computational methods for predicting protein interactions of natural products. Comput Struct Biotechnol J. 2019, 28(17):1367-1376. doi: https://doi.org/10.1016/j.csbj.2019.08.008. PMID: 31762960
About the Author
Dr. Aurélien F. A. Moumbock is currently a postdoc at the University of Freiburg (Germany), where he obtained a PhD in Pharmaceutical Bioinformatics in 2022. He holds both B.Sc. and M.Sc. degrees in Chemistry from the University of Buea (Cameroon).
"Macromolecular Engineering to Potentiate Antibiotics against Gram-Negative Superbugs and Eradicate Mixed Species Biofilms In Vivo"
We developed new antimicrobial polymers following a simple synthetic strategy. The biocompatible polymers showed excellent potency to rejuvenate resistant antibiotics against Gram-negative superbugs. Most importantly, in combination with the antibiotics this macromolecule eradicates multi-species biofilm infections established in a murine model.
Macromolecular Engineering to Potentiate Antibiotics against Gram-Negative Superbugs and Eradicate Mixed Species Biofilms In Vivo
Sudip Mukherjee, Jayanta Haldar
The increasing prevalence of multi drug resistant Gram-negative pathogens and declining approval of novel antibiotics has created a huge void in the clinical pipeline. In addition to these, infections associated with multi species superbugs and their concomitant existences in mixed species biofilms are refractory to all classes of antibiotics. To tackle this drug resistance in Gram-negative bacteria and to cure the infections caused by multispecies biofilms, the present study showcases engineered amphiphilic cationic macromolecules to rehabilitate and repurpose the resistant antibiotics like rifampicin and fusidic acid. This membrane-perturbing molecule hampers the bacterial efflux machinery thereby showing elevated uptake of these antibiotics. The macromolecule-antibiotic cocktails disrupted preformed Gram-negative bacterial biofilms. The cocktail also encased compelling efficiency to disrupt polymicrobial biofilms composed of P. aeruginosa and MRSA, a critical microbial niche abundant in various fatal infections. Significantly, the antibiotic in combination with our engineered macromolecule was biocompatible, with an excellent in-vivo antibacterial efficacy (> 99% reduction) against multi-microbial biofilm established in mice. In the era of void in the antibiotic arsenal, these results promise tremendous potential of our macromolecule to potentiate antibiotics and tackle the rising threat of antimicrobial resistance.
About the Author
Sudip Mukherjee is an Integrated Ph.D. student under the guidance of Prof. Jayanta Haldar. My research at JNCASR deals with the development of novel antimicrobial polymeric materials. I am always up for different sports and a die heart Madridista.
Vandana Nandakumar – 61
Bharathiar University
"SYNTHESIS AND EVALUATION OF ANTI-CANCER ACTIVITY OF NITROQUINOLONE BASED ACYLHYDRAZONES AGAINST HUMAN LUNG CANCER A549 CELL LINES"
SYNTHESIS AND EVALUATION OF ANTI-CANCER ACTIVITY OF NITROQUINOLONE BASED ACYLHYDRAZONES AGAINST HUMAN LUNG CANCER A549 CELL LINES
**Corresponding Author, mail id: suresh@buc.edu.in
1Department of Chemistry, Bharathiar University, Coimbatore, TamilNadu, India, 641046
Quinoline-based natural and synthetic compounds have privileged position in medicinal chemistry due to their diverse biological activities and this moiety is frequently used in novel drug design. Hydrazide analogues of various heterocycles have proven to exhibit antidepressant, anticonvulsant and anti-inflammatory behavior. Therefore fusion of aromatic hydrazide sidechain with quinoline may lead to several bio-active derivatives. Nitro substituted aromatic ring systems have been used in antifungal and anti-inflammatory drugs. Functionalising quinoline core with a nitro group could widen pharmaceutical importance due to hydrogen bonding interaction between Nitro group and ring nitrogen, which could stabilize the quinoline core and enhance the active sites at the angular position.
In this work, new series of 8-nitroquinolone based aromatic heterocyclic acyl hydrazones have been synthesised and characterised through various spectroscopic techniques. They have been evaluated for their anti-lung cancer activity against A459 cell lines through MTT assay to determine cytotoxicity. The best active compounds in the series were chosen for further detection of apoptosis through florescence microscopic techniques using AO/EtBr and DAPI. The nuclear fragmentation during the cell cycle was studied through flow cytometric techniques. The results indicate that the compounds possess good anticancer property due to the cell cycle arrest at the S-Phase. Molecular docking with the BCL-2 protein indicate the possible modes of interaction of the synthesised compounds were in comparison with that of the standard.
References:
Ajani, Olayinka O., King T. Iyaye, and Olabisi T. Ademosun. “Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs–a review.” RSC advances29 (2022): 18594-18614.
Narasimhamurthy, Kereyagalahally H., et al. “Synthesis of bioactive quinoline acting as anticancer agents and their mode of action using in silico analysis towards Aurora kinase A inhibitors.” Chemical Data Collections35 (2021): 100768.
Koçyiğit, Ümit Muhammet, et al. “Arylated quinoline and tetrahydroquinolines: Synthesis, characterization and their metabolic enzyme inhibitory and antimicrobial activities.” ChemistrySelect37 (2022): e202203469.
Yang, Guan-Zhou, et al. “Design, synthesis, and antifungal evaluation of novel quinoline derivatives inspired from natural quinine alkaloids.” Journal of agricultural and food chemistry41 (2019): 11340-11353.
Altwaijry, Najla, et al. “In Vitro and In Vivo Antitumor Activity of Indolo [2, 3-b] Quinolines, Natural Product Analogs from Neocryptolepine Alkaloid.” Molecules3 (2021): 754.
Panda, Pravati, and Subhendu Chakroborty. “Navigating the synthesis of quinoline hybrid molecules as promising anticancer agents.” ChemistrySelect33 (2020): 10187-10199.
Anduran, Emilie, et al. “Hypoxia-activated prodrug derivatives of anti-cancer drugs: A patent review 2006–2021.” Expert Opinion on Therapeutic Patents1 (2022): 1-12.
Liang, Dinghua, et al. “Evaluation of nitrobenzyl derivatives of camptothecin as anti-cancer agents and potential hypoxia targeting prodrugs.” Molecules8 (2018): 2041.
Nepali, Kunal, Hsueh-Yun Lee, and Jing-Ping Liou. “Nitro-group-containing drugs.” Journal of medicinal chemistry6 (2018): 2851-2893.
Adams, G. E., and I. J. Stratford. “Hypoxia Mediated Nitro Heterocyclic Drugs in The Radio and Chemotherapy of Cancer.” Biochemical Pharmacology1 (2013): 71.
Verma, Sangeeta, et al. “Quinoline Hydrazide/Hydrazone Derivatives: Recent Insights on Antibacterial Activity and Mechanism of Action.” ChemMedChem(2023).
Liu, Xinjin, et al. “Discovery of Aryl Benzoyl Hydrazide Derivatives as Novel Potent Broad-Spectrum Inhibitors of Influenza A Virus RNA-Dependent RNA Polymerase (RdRp).” Journal of Medicinal Chemistry5 (2022): 3814-3832.
Yue, Kairui, et al. “First-in-Class Hydrazide-Based HDAC6 Selective Inhibitor with Potent Oral Anti-Inflammatory Activity by Attenuating NLRP3 Inflammasome Activation.” Journal of Medicinal Chemistry18 (2022): 12140-12162.
Kamat, Vinuta, et al. “Pyridine-and thiazole-based hydrazides with promising anti-inflammatory and antimicrobial activities along with their in silico studies.” ACS omega39 (2020): 25228-25239.
Yan, Xiao-Jing, et al. “A chromone hydrazide Schiff base fluorescence probe with high selectivity and sensitivity for the detection and discrimination of human serum albumin (HSA) and bovine serum albumin (BSA).” Journal of Photochemistry and Photobiology A: Chemistry422 (2022): 113576.
Haroon, Muhammad, et al. “Synthesized thiazole-based hydrazides and their spectral characterization along with biological studies: Promising quantum chemical insights.” Journal of Molecular Structure1270 (2022): 133923.
Mohamed, Shaaban K., et al. “Insight into the Crystal Structures and Potential of Two Newly Synthesized Naproxen-Based Hydrazide Derivatives as Potent COX-2 Inhibitors.” Applied Biochemistry and Biotechnology12 (2022): 5781-5807.
Coşar, Ebru Didem, et al. “Anticholinesterase activities of novel indole-based hydrazide-hydrazone derivatives: design, synthesis, biological evaluation, molecular docking study and in silico ADME prediction.” Journal of Molecular Structure1247 (2022): 131398.
Shyamsivappan, Selvaraj, et al. “Synthesis and X-ray study of dispiro 8-nitroquinolone analogues and their cytotoxic properties against human cervical cancer HeLa cells.” Medchemcomm3 (2019): 439-449.
Calvin Omolo
United States International University - Africa
"Dual acting acid-cleavable self-assembling prodrug from hyaluronic acid and ciprofloxacin: A Biomimetic system for simultaneously targeting bacterial infections and cancer"
Dual acting acid-cleavable self-assembling prodrug from hyaluronic acid and ciprofloxacin: A Biomimetic system for simultaneously targeting bacterial infections and cancer
Usri H. Ibrahim, Nikita Devnarain, Calvin A. Omolo, Rene Khan, Thirumala Govender
The high risks of infection in cancer patients are attributed to compromised host defenses and the sequelae of treatment. This has prompted many research efforts to develop new strategies with dual activity in treating cancer while simultaneously working against bacterial infections. Therefore, this study aimed to synthesize a novel acid-cleavable hyaluronic acid-based prodrug of ciprofloxacin (HA-Cip) to enhance its anticancer potential and biosafety while maintaining its antibacterial activity. HA-Cip was synthesized using a multi-step reaction scheme and products from each step were fully characterized via FT-IR, HR-MS, and H1 NMR techniques. Self-assembly of HA-Cip prodrug obtained stable micelles with an average particle size of 237.89 ± 25.74 nm, PDI of 0.265 ± 0.013 and ZP of -17.82 ± 1.53 mV at normal physiological pH 7.4, which destabilize and rearrange in response to acidic pH change to 6.0 with average particle size, PDI and ZP of 179.27 ± 7.66 nm, 0.517 ± 0.026 and -8.31 ± 1.26 mV, respectively. Cytotoxicity studies against human kidney (HEK293) cells showed excellent biosafety properties with more than 80% cell viability, and haemolysis studies revealed advanced biosafety with less than 0.3% haemolysis activity. Ciprofloxacin (Cip) release from HA-Cip prodrug was faster at pH 6.0 with 100% cumulative drug release after 5 hours compared to normal physiological pH with about 90% after 24 hours. HA-Cip showed enhanced in vitro antibacterial activity against E. coli and K. pneumoniae at pH 6.0 compared to bare Cip and comparable MIC values at pH 7.4. In addition, a 5.4-fold enhancement in Cip anticancer activity against HepG2 cells was achieved by HA-Cip via improvement of the Cip oxidative stress induction ability. Furthermore, HA-Cip showed cytoprotective activity through inhibition of cell death in HEK293 cells, which may contribute to the reduction of side effects of Cip on non-cancerous cells. Our findings propose HA-Cip prodrug for effective targeted delivery of Cip to treat both bacterial infections and cancer.
Philipp Otten – 39
WWU Muenster (University of Muenster)
"Fragment-based design of mycobacterial thioredoxin reductase inhibitors by socializing a fragment and exploring a vast virtual chemical space"
The fragment-based design and synthesis of a novel and potent inhibitor of the mycobacterial thioredoxin reductase utilising AI-driven synthesis predictions.
Fragment-based design of mycobacterial thioredoxin reductase inhibitors by socializing a fragment and exploring a vast virtual chemical space
Institute of Pharmaceutical and Medicinal Chemistry, University of Münster, Corrensstr. 48, 48149 Münster, Germany 2 German Center of Infection Research, University of Münster, Corrensstr. 48, 48149 Münster, German
Institute of Biochemistry, University of Münster, 36, 48149 Münster, German
European Institute for Molecular Imaging (EIMI), 15, 48149 Münster, Germany
Tuberculosis still is one of the deadliest infectious diseases worldwide and the rise in multi-resistant strains is of great concern. A novel target in M. tuberculosis is the thioredoxin reductase (TrxR) which is essential for survival. differs significantly from its human counterpart and was already shown to be druggable [1&2]. Based on an exhaustive crystallographic fragment screening a promising fragment binding in the NADPH-binding pocket was evolved in a fragment-based design approach combining structure-based design and computational strategies with AI-powered synthesis predictions and molecular docking. Unfortunately, this fragment is “unsociable” as the necessary attachment vectors for growth are unavailable and no analogues are commercially available [3]. First, the fragment was “socialized” via scaffold-hopping allowing growth into the binding pocket. Afterward, multiple derivatives of the socialized fragment were designed by combining combinatorial fragment-growing strategies with AI-powered synthesis predictions and docking. The IBMRXN [4] and ASKCOS [5] suites were used to decide which compounds are synthetically feasible and determine the best reaction conditions for each reaction. The syntheses based on these predictions led to a compound that shows potent inhibition of the TrxR. Together with the obtained complex structure of this hit, a new starting point for the development of bioactive TrxR inhibitors was identified.
References:
1 Lin, K. et al.: PLoS pathog., 2016 (12), e1005675
2 Koch, O. et al.: J. Med. Chem., 2013(56), 4849
3 St. Denis, J. D. et al.: RSC Med. Chem., 2021(12), 321-32
4 Schwaller, P. et al.: Chem. Sci., 2018(9), 6091-6098
5 Gao, H. et al.: ACS Cent. Sci. 2018(11), 1465–1476
About the Author
After receiving my Master’s Degree in Pharmaceutical Sciences, I continue to work in Prof. Oliver Koch’s group dealing with the combination of in-silico, data-driven methods, fragment-based drug design and “traditional” synthesis.
"Investigations on structural diversity and biosynthesis of biarylitides – a novel class of minimal RiPPs"
The smallest gene ever described plays a decisive role in the biosynthesis of biarylitides - a novel class of minimal RiPPs
Investigations on structural diversity and biosynthesis of biarylitides – a novel class of minimal RiPPs
Leo Padva, Maria Guadalupe Soto Zarazua, Max Crüsemann
University of Bonn, Institute of Pharmaceutical Biology, Nussallee 6, 53115 Bonn, Germany
A recently discovered, novel class of ribosomally-synthesized and post-translationally modified peptides (RiPPs) are the biarylitides.[1] In their biosynthesis, the smallest coding gene ever described across the tree of life plays a decisive role. This 18 bp gene encodes a precursor pentapeptide, which is enzymatically modified and cleaved into a tripeptide with an unusual biaryl bond linking two aromatic amino acids. This biaryl bond is generated by a downstream-encoded, specific cytochrome P450 monooxygenase. The corresponding minimal biosynthetic gene cluster (BGC) was discovered in a strain of the less explored actinobacterial genus Planomonospora. Global bioinformatic analyses revealed biarylitide BGCs in at least 200 bacterial genomes, encoding further modifying enzymes in some cases, indicating a large, undiscovered structural diversity of biarylitides. This project focuses on the isolation and characterisation of structurally modified biarylitides, which is aimed to be achieved by the heterologous expression of selected BGCs. We report the detection of six novel biarylitides, three of which appear to be the first modified members described. Additionally, systematic mutation experiments of the biarylitide precursor gene resulted in the heterologous production of peptides with altered sequences. Among these is the first in vivo produced biarylitide with tryptophan at the crosslinking position.
References:
Zdouc MM et al. (2021) Cell Chem Biol 28 (5), 733-739
Victor Santos de Paiva – 41
University of Coimbra
"Estimation of drug plasma protein binding using isothermal titration calorimetry and binding to albumin hydrogels"
New methodologies to estimate the fraction bound of drugs with albumin, and the physicochemical characterization of the interactions.
Estimation of drug plasma protein binding using isothermal titration calorimetry and binding to albumin hydrogels.
Victor S. Paiva, Zaida L. Almeida, Pedro F. Cruz, Jaime Samelo, Daniela C. Vaz, Rui M. M. Brito, Maria João Moreno
Coimbra Chemistry Center – Institute of Molecular Sciences (CQC-IMS), Department of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal
Plasma protein binding (PPB) has a strong effect on drug pharmacokinetics, since it is usually the free drug that permeates the endothelia towards the therapeutic target. Equilibrium dialysis is the “gold standard” methodology to measure PPB. However, this approach is time consuming, and binding of the drugs to the dialysis membrane may lead to artifacts1. The demand for PPB data on an always increasing number of compounds has stimulated attempts to streamline existing methodology, as well as to develop new procedures2. Protein hydrogels are of particular interest due to their high water content, which imposes low conformational constraints to the protein, and facilitates the drug diffusion and the rapid equilibrium between the aqueous environment and the protein3.
In this work, we use BSA-hydrogels, recently developed by us3, in order to characterize the PPB of a large number of drugs. A good correlation was observed with PPB data obtained by standard approaches. To characterize the thermodynamics and kinetics of BSA-drug interactions for a small set of drugs, ITC and NMR4 were also used.
References:
A. Smith, L. Di, E.H. Kerns, The effect of plasma protein binding or in vivoefficacy: misconceptions in drug discovery, Nat. Rev. Drug Discov., 2010, 9, 929–939.
G.L. Trainor, The importance of plasma protein binding in drug discovery, Expert Opin. Drug Discov., 2007, 2, 51–64.
Coelho, C.D.F.; Jesus, J.A.; Vaz, D.C.; Lagoa, R.; Moreno, M.J. BSA-PEG Hydrogel: A Novel Protein-Ligand Binding 3D Matrix. Sci. Forum 2022, 11, 1.
Gallo, S. Matteucci, N. Alaimo, E. Pitti, M. V. Orsale, V. Summa, D. O. Cicero, E. Monteagudo, A novel method using nuclear magnetic resonance for plasma protein binding assessment in drug discovery programs, Journal of Pharmaceutical and Biomedical Analysis, 2019, Volume 167, Pages 21-29.
About the Author
Victor S. Paiva studied chemistry in the University of Brasília. For his master’s degree he is currently investigating the interactions of albumin and drugs, in order to characterize the system thermodynamic and kinetic.
"Design, Synthesis and Evaluation of Heterocyclic Analogs as Dual AChE and PDE4 Inhibitors for Alzheimer's Treatment"
Design, Synthesis and Evaluation of Heterocyclic Analogs as Dual AChE and PDE4 Inhibitors for Alzheimer’s Treatment
Vaishali M. Patil
Department of Pharmaceutical Chemistry, KIET School of Pharmacy, KIET Group of Institutions, Delhi-NCR, Ghaziabad, Uttar Pradesh, India
Recent reports highlights role of multifunctional anti-Alzheimer’s agents targeting different pathways/enzymes. AChE and PDE4 inhibitors have displayed significant improvement in cognitive function. In the present study design of few heterocyclic imidazo [1,2-α] pyridine analogs of and their synthesis is reported. These analogs were evaluated as dual acetylcholinesterase (AChE) and phosphodiesterase 4 (PDE4) inhibitors for Alzheimer’s treatment using Donepezil as a standard drug. The results showing activity at micromolar (µM) range by synthesized analogs was analyzed for comparing selective against the selected targets and neuroprotective effects. Compound 5E displayed comparable inhibition of AChE with Donepezil in the hippocampus of mice model. Structure activity relationship analyses concluded role of various functional groups and their position for dual inhibitory activity.
"Protein kinase inhibitor ceritinib blocks ectonucleotidase CD39 – a promising target for cancer immunotherapy"
Protein kinase inhibitor ceritinib blocks ectonucleotidase CD39 – a promising target for cancer immunotherapy
Carolin S. Pikullik, Laura Schäkel, Salahuddin Mirza, Vittoria Lopez, Riham Idris, Haneen Al-Hroub, Riekje Winzer, Eva Tolosa, Christa E. Müller
Pharmaceutical & Medicinal Chemistry, PharmaCenter Bonn, Pharmaceutical Institute, Rheinische Friedrich-Wilhelms-Universität Bonn, An der Immenburg 4, D-53121 Bonn, Germany.
E-mail: carolin.pikullik@uni-bonn.de, christa.mueller@uni-bonn.de
Accumulation of extracellular adenosine has emerged as a key mechanism of immune escape. Adenosine represents one of the strongest physiological immunosuppressive mediators, but also promotes tumor cell proliferation, metastasis and angiogenesis.1,2 Ectonucleotidases, which catalyze the hydrolysis of ATP to AMP (CD39) and further to adenosine (CD73), are upregulated on many cancer cells. Thus, these enzymes are considered as promising targets for the (immuno)therapy of cancer.3 However, small molecule CD39 inhibitors suitable for therapeutic applications are lacking. Since most kinase inhibitors used in cancer therapy target the (intracellular) binding site for the co-factor ATP on protein kinases, we figured that some of them might additionally interact with the (extracellular) ATP substrate binding site of CD39. Thus, we screened 50 approved ATP-competitive protein kinase inhibitors on human CD39. Ceritinib, an anaplastic lymphoma kinase (ALK) inhibitor, was discovered to additionally inhibit CD39 at a therapeutically relevant concentration.4 The drug displays a non-competitive, allosteric mechanism of inhibition (Ki = 11.0 µM). Furthermore, Ceritinib features high metabolic stability, optimized physicochemical properties and brain permeation and is therefore an ideal starting point to develop more potent and selective CD39 inhibitors. We currently optimize the ceritinib scaffold for CD39 inhibition and/or dual CD39/ALK inhibition.
References:
Allard, B. et al. Curr. Opin. Pharmacol. 2016, 29, 7-16. [2] Borodovsky, A. et al. J. Immunother. Cancer 2020, 8, e000417. [3] Timerpi, E. et al. Int. J. Mol. Sci.2021, 22, 8068. [4] Schäkel, L. et al. J. Immunother. Cancer2022, 10, e0046660.
Thanigaimalai Pillaiyar – 44
Institute of Pharmacy, Eberhard Karls University Tübingen
"Thioesters as Novel SARS-CoV-2 Main Protease Inhibitors: Design, Synthesis, Enzyme Inhibition, Structure-Activity Relationship, Antiviral Activity, and X-Ray Structure Determination"
We designed, synthesized, and evaluated a series of novel small molecule thioesters as SARS-CoV-2 Mpro inhibitors, which led to promising inhibitors with IC50 values of around 10 nM. These inhibitors also showed promising antiviral efficacy without cytotoxicity. They could be used to treat a broader range of coronavirus infections.
Thioesters as Novel SARS-CoV-2 Main Protease Inhibitors: Design, Synthesis, Enzyme Inhibition, Structure-Activity Relationship, Antiviral Activity, and X-Ray Structure Determination
Thanigaimalai Pillaiyar,*1 Philipp Flury,1 Nadine Krüger,2 Haixia Su,3 Laura Schäkel,4 Elany Barbosa Da Silva,5 Olga Eppler,1 Thales Kronenberger,1,6 Tianqing Nie,3 Stephanie Luedtke,5 Cheila Rocha,2 Katharina Sylvester,4 Marvin R.I. Petry,4 Antti Poso,1,7 Stefan Pöhlmann,2,8 Michael Gütschow,4 Anthony J. O’Donoghue,5 Yechun Xu,3 Christa E. Müller,4 and Stefan Laufer1
Institute of Pharmacy, Pharmaceutical/Medicinal Chemistry and Tübingen Center for Academic Drug Discovery, Eberhard Karls University Tübingen, Auf der Morgenstelle 8, 72076 Tübingen, Germany. Cluster of Excellence iFIT (EXC 2180) ‘Image-Guided & Functionally Instructed Tumor Therapies’, University of Tübingen, Tübingen72076, Germany;
Infection Biology Unit, German Primate Center, Leibniz Institute for Primate Research Göttingen, Kellnerweg 4, 37077 Göttingen, Germany;
CAS Key Laboratory of Receptor Research, and Stake Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
PharmaCenter Bonn, Pharmaceutical Institute, Pharmaceutical & Medicinal Chemistry, University of Bonn, An der Immenburg 4, D-53121 Bonn;
Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California, United States of America;
Department of Medical Oncology and Pneumology, Internal Medicine VIII, University Hospital Tübingen, Otfried-Müller-Str. 14, Tübingen, 72076, Germany;
School of Pharmacy, Faculty of Health Sciences, University of Eastern Finland, 70211 Kuopio, Finland;
In coronaviruses, the main protease (Mpro, 3CLpro) is a validated target due to its crucial function in viral replication and transcription [1]. Based on previous work [2], we designed, synthesized, and evaluated a series of novel small molecule thioesters as SARS-CoV-2 Mpro inhibitors [3], which led to promising inhibitors with IC50 values of around 10 nM. In cell- based experiments, these inhibitors also showed promising antiviral efficacy without cytotoxicity. Co-crystallization of selected compounds with the SARS-CoV-2 Mpro was accomplished to gain insights into the molecular interactions, and crystal structures were established to validate the covalent bond formation of inhibitors with the protease. The discovery that strong Mpro inhibitors of SARS-CoV-2 also inhibited other beta-coronaviruses, such as SARS-CoV-1 and MERS-CoV, with comparable efficacy could be used to treat a broader range of coronavirus infections.
Keywords: Anti-viral therapeutics; COVID-19; Inhibitors; Main protease (Mpro); SARS-CoV- 2.
References:
Pillaiyar, T. et al. Med. Res. Rev. 2021, 41, 72-135. [2] Breidenbach, J. et al. Angew. Chem. 2021, 133, 10515-10521. [3] Pillaiyar, T. et al. J. Med. Chem. 2022, 65, 9376–9395.
About the Author
Dr. Pillaiyar establishes a research group at the Tuebingen Center for Academic Drug Discovery, University of Tubingen. His research interests include the medicinal chemistry of GPCRs and SARS-CoV-2 Mpro-inhibitors.
René Richarz – 45
University of Bonn
"In situ modification of FR900359 biosynthesis in Chromobacterium vaccinii MWU205"
In situ modification of FR900359 biosynthesis in Chromobacterium vaccinii MWU205
René Richarz*, Christoph Ulbricht, Gabriele M. König and Max Crüsemann+
Institute of Pharmaceutical Biology, Rheinische Friedrich-Wilhelms-Universität Bonn, Nussallee 6, 53115 Bonn, Germany. + E-Mail: mcruesem@uni-bonn.de
FR900359 (FR) (Figure 1) is an eight-membered, cyclic depsipeptide, which belongs to a small and specialized group of natural products named chromodepsins. So far, FR production has been reported from two different bacteria, the obligate plant symbiont “Candidatus Burkholderia crenata” and the free-living Chromobacterium vaccinii MWU205. In both cases a non-ribosomal peptide synthetase (NRPS) machinery is responsible for its assembly.1,2
FR has proven to be an excellent tool for the investigation of Gq protein-coupled signaling pathways, that regulate important cellular processes in diverse organisms.3 This activity also makes it a potential drug lead for the treatment of diseases, such as asthma or cancer.1 Due to these interesting features there is a strong desire for new FR derivatives with altered activities. This includes the targeting of signaling pathways regulated by other G protein family members as well as the installation of chemical groups for the selective modification of the FR scaffold.
Here we report our first results on the in situ modification of FR biosynthesis by C.vaccinii to enhance the production of FR as well as natural occurring FR derivatives and to generate altered FR molecules.
Figure 1: Structure of FR900359
References:
C. Hermes, G. M. König and M. Crüsemann, Nat. Prod. Rep., 2021, 38, 2276-2292.
C. Hermes, R. Richarz, D. A. Wirtz, J. Patt, W. Hanke, S. Kehraus, J. H. Voss, Küppers, T. Ohbayashi, V. Namasivayam, J. Alenfelder, A. Asuka Inoue, Mergaert, M. Gütschow, E. Kostenis, G. M. König and M. Crüsemann, Nat. Commun., 2021, 12, 144.
R. Schrage, A.-L. Schmitz, E. Gaffal, S. Annala, S. Kehraus, D. K. Wenzel, G. M. König and E. Kostenis, Nat. Commun., 2015, 6, 10156.
Detection of protease activity in solution has a wide range of applications, including determining their presence as contaminants in clinical and research samples. At Detact Diagnostics we developed ProTact to efficiently and easily detect the presence of proteases at low concentrations.
Residual protease activity in research samples is a major source of variability in processing and analysis. Whether the protease origin is the sample itself, as in the case of cell lysates, or external, from contamination, a quick and effortless way to determine the presence of protein-digesting enzymes is useful and advisable. At Detact Diagnostics we have developed a near-infrared (NIR) fluorescence assay that can detect protease presence in solution with high sensitivity and fast response (in the order of minutes) allowing the screening of a large number of samples in a 96-well plate format. Alternatively, using the DeNIRO® device, single samples can be analysed in batch, making it extremely appealing for monitoring samples at progressive steps of processing or purification. On top of that, the assay can detect a wide range of bacterial proteases having different reaction mechanisms, such as serine and metalloproteases.
About the Author
Simone Savino is an experienced structural biologist and biochemist who is now part of the Detact Diagnostics team, which is focused on medical devices and bacterial proteases detection in a variety of samples.
"Design and synthesis of novel P2Y2 receptor antagonists"
Design and synthesis of novel P2Y2 receptor antagonists
Schlegel, K.1; Junker, A.1
European Institute for Molecular Imaging (EIMI), University of Münster, Waldeyerstraße 15, 48149 Muenster, Germany
P2Y2 receptors (P2Y2R) belong to the P2Y1-like Gq/11-coupled subfamily of purinergic P2Y receptors [1]. Pancreatic ductal adenocarcinoma (PDAC), one of the most aggressive cancer types worldwide, is associated with an upregulation of the P2Y2R expression [2]. Despite many efforts, there is still no effective drug for the treatment of PDAC available, resulting in a poor prognosis for patients. P2Y2R antagonists could offer a novel approach to the successful treatment of PDAC. AR-C118925 (Figure 1, left) is a P2Y2R antagonist that displays high P2Y2R activity but suffers from low oral availability [3]. Therefore, taking AR-C118925 as a lead structure, we decided to explore structural variations in two different areas of the molecule (Figure 1) to develop P2Y2R antagonists with improved oral bioavailability and metabolic stability.
Figure 1: Lead compound AR-C118925 (left) and novel P2Y2R antagonists 1-9.
References:
Rafehi, M.; Burbiel, J. C.; Attah, I. Y.; Abdelrahman, A.; Müller, C. E., Signal. 2017, 13, 89-103.
Hu, L. P.; Zhang, X. X.; Jiang, S. H.; Tao, L. Y.; Li, Q.; Zhu, L. L.; Yang, M. W.; Huo, Y. M.; Jiang, Y. S.; Tian, G. A.; Cao, X. Y.; Zhang, L.; Yang, Q.; Yang, X. M.; Wang, Y. H.; Li, J.; Xiao, G. G.; Sun, Y. W.; Zhang, Z. G., Clin Cancer Res 2019, 25, 1318-1330.
"A small fluorescent ligand to investigate soluble epoxide hydrolase engagement in living cells"
A small fluorescent ligand to investigate soluble epoxide hydrolase engagement in living cells
Julia Schoenfeld, DE, Steffen Brunst, DE, Luisa Burgers, DE, Peter Breunig, DE, Murphy DeMeglio, DE, Felix F. Lillich, DE, Lilia Weizel, DE, Robert Fürst, DE, Jasmin Hefendehl, DE, Ewgenij Proschak, DE, Kerstin Hiesinger, DE
Soluble epoxide hydrolase (sEH) is ubiquituos in the human organism and metabolizes the highly biologically active epoxy-fatty acids (EETs) to the less active corresponding diols. Since sEH metabolism is the main pathway for the deactivation of EETs it represents a promising target for a variety of diseases. [1] In disease models of central nervous system disorders such as Alzheimer´s or Parkinson´s disease sEH upregulation was observed while inhibition of sEH resulted in neuroprotective effects. [2] In this work, we present the design and synthesis of the small fluorescent sEH ligand JSF68. Starting from the potent sEH inhibitor GSK2256294A we replaced the triazine moiety by the small fluorophore nitrobenzoxadiazole (NBD). The resulting fluorescent sEH ligand displayed excellent potency in vitro (IC50 < 1.5 nM in our enzyme activity assay) and was suitable for fluorescence polarization measurements. We demonstrate the applicability of JSF68 in a NanoBRET-based assay which is the first sEH target engagement assay in living cells reported so far. Furthermore, the fluorescent ligand was used to detect sEH in sEH-transfected HEK293T cells and in primary mouse astrocytes by fluorescence microscopy.
References:
M. Pallàs, S. Vázquez, C. Sanfeliu, C. Galdeano, C. Griñán-Ferré, Biomolecules,2020.
"Inhibition of thermal and shear-induced aggregation of albumin by Centella asiatica extract: A mechanistic insight"
Inhibition of thermal and shear-induced aggregation of albumin by Centella asiatica extract: A mechanistic insight
Laipubam Gayatri Sharma1, Chinmaya Panda1 and Lalit Mohan Pandey1
Biointerface and Environmental Engineering Lab, Indian Institute of Technology, Guwahati, 781039, India.
The aggregation of proteins and peptides has emerged as a significant cause of neurodegeneration and loss of formulation integrity in pharmaceutical product development 1. Various natural sources are being explored for inhibiting protein aggregation. Centella asiatica (CA) is a plant known for its neuroprotection property and memory enhancement. Antioxidative, radical scavenging, and modulation of cholinergic activities are some properties attributed to the neuroprotection ability of the plant 2. Components of CA could likely bind to proteins and peptides owing to their small size and aid the native structure stabilization when exposed to various external stimuli. Therefore, the effect of CA extract against thermal and shear-induced aggregation of bovine serum albumin (BSA) at 60°C, and a shear rate of 300 s-1 was evaluated. Based on increasing solvent polarity, three solvents (viz., ethyl acetate, methanolic, and water) were used for generating CA extract. The aggregation was monitored through Thioflavin T assay, dynamic light scattering, circular dichroism, and atomic force microscopy. The interaction of the extract and BSA was thermodynamically investigated through isothermal titration calorimetry. In vitro toxicity of the aggregates and the CA extract-mediated cytotoxicity rescue were also evaluated. This study found that the water and ethyl acetate extract significantly inhibit aggregation.
Keywords: Protein aggregation; Centella asiatica; Shear; Thermal; BSA
References:
Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat. Neurosci.2018, 21 (10), 1332–1340. https://doi.org/10.1038/s41593-018-0235-9.
Wong, J. H.; Barron, A. M.; Abdullah, J. M. Mitoprotective Effects of Centella Asiatica (L.) Urb.: Anti-Inflammatory and Neuroprotective Opportunities in Neurodegenerative Disease. Front. Pharmacol.2021, 12, 687935. https://doi.org/10.3389/fphar.2021.687935.
Sabrina Silva Mendonça – 49
Universidade Federal de Goiás
"Cheminformatics-aided discovery of new chemotypes for multistage activity antimalarials"
Were found inhibitors of plasmodium life cycle, selectec through computational methodologies and experimental validation. The next steps will be sumitt the best compounds to enzimatic assay.
Cheminformatics-aided discovery of new chemotypes for multistage activity antimalarials
Mendonça, S. S.1; Magalhães, M. L.2; Alvarez, L. C. S.2; Calit, J. P.3; Ferreira, L. T.2; Cassiano, G. C.4; Moreira-Filho, J. T.1; Borba, J. V. B.1; Neves, B. J.1; Eastman, R. T.5; Costa, F. T. M.2; Bargieri, D. Y.3; Andrade, C. H.1
Laboratory of Molecular Modeling and Drug Design (LabMol), Faculty of Pharmacy, Universidade Federal de Goiás, Goiânia, 74605-170, GO, Brazil.
Laboratory of Tropical Diseases (LDT) – Prof. Dr. Luiz Jacinto da Silva, Department of Genetics Evolution, Microbiology and Immunology, Institute of Biology, Universidade de Campinas (UNICAMP), 13083-970, Campinas, SP (Brazil).
Institute of Biomedical Sciences, University of São Paulo, São Paulo, SP, 05508-000, Brazil.
Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical, Universidade Nova de Lisboa, 1099-085, Lisboa, Portugal.
Laboratory of Malaria and Vector Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA.
Due to the emergence of artemisinin-resistant in Africa[1], the search for new antimalarials with novel mechanisms of action, such as transmission-blocking activity, is urgent. The aim of this study was to develop and validate cheminformatics approaches for discovering new antimalarial compounds with activity against asexual and sexual stages of Plasmodium spp. Initially, shape-based models for inhibitors of aurora kinase-2 from P. falciparum (PfArk-2) (AUC=0.97, EF=9, BEDROC=0.69), as well QSAR models for ookinete formation inhibition (LigthGBM-ECFP4+Mordred: Bal-ACC=0.8, Se=0.9, and Sp=0.7), were generated. These models were employed in a virtual screening to find compounds with activity in PfArk-2, 3D7 and W2 (models previously developed), and ookinete formation inhibition. Moreover, we also excluded compounds that were with oral acute toxicity[2] and hERG blockage potential[3], resulted in ten compounds prioritized for experimental validation. Four of them demonstrated activity against strains 3D7 and Dd2 P. falciparum at concentrations of 0.11–0.27µM, and selectivity indices ranging at 17-66. Moreover, at 10µM, three compounds inhibited ookinete formation of P. berghei. For conclusion, the cheminformatics approaches used in this study succeeded to predict multi-stage compounds, with good potency. The best compounds will be submitted to enzymatic assays in PfArk-2, and could represent new chemical scaffolds for hit-to-lead optimization.
References:
Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O. T.; Tachibana, S.-I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D. A.; Kimura, E.; Palacpac, N. M. Q.; Odongo-Aginya, E. I.; Ogwang, M.; Horii, T.; Mita, T. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med.2021, 385, 1163–1171.
Borba, J. V. B.; Alves, V. M.; Braga, R. C.; Korn, D. R.; Overdahl, K.; Silva, A. C.; Hall, S. U. S.; Overdahl, E.; Kleinstreuer, N.; Strickland, J.; Allen, D.; Andrade, C. H.; Muratov, E. N.; Tropsha, A. STopTox: An in Silico Alternative to Animal Testing for Acute Systemic and Topical Toxicity. Environ. Health Perspect.2022, 130.
Braga, R. C.; Alves, V. M.; Silva, M. F. B.; Muratov, E.; Fourches, D.; Lião, L. M.; Tropsha, A.; Andrade, C. H. Pred-hERG: A Novel web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform.2015, 34, 698–701.
About the Author
PhD student in Tropical Medicine and Public Health, at Federal University of Goiás, Brazil. Focusing in apply computational methods (bioinformatics and chqmoinformatics) to drug discover new treatments for emergence/tropical diseases.
"Impacts of DNA repair on anticancer drugs: synthesis and activity of novel acylfulvene analogs with aromatic groups"
Synthetical lethality approach of targeting cancer by using a DNA alkylating agents with subsequent repair inhibition.
Impacts of DNA repair on anticancer drugs: synthesis and activity of novel acylfulvene analogs with aromatic groups
Laura Slappendel1, Vakil Takhaveev1, Claudia M. N. Aloisi2, Laura P. Brandt3, Mark Rubin3, Orlando S. Schärer4, Shana J. Sturla1
Department of Health Sciences and Technology, ETH Zürich, Zürich, Switzerland
Institut de Biologie de l’École Normale Supérieure, Paris
Bern Center for Precision Medicine and Department for Biomedical Research, Bern, Switzerland
Center for Genomic Integrity, Institute for Basic Science, Ulsan, 44919 Republic of Korea; Department of Biological Sciences *E-mail: laura.slappendel@hest.ethz.ch
Despite their wide use in cancer therapy, the efficacy and safety of DNA alkylating anticancer agents are hindered by cellular mechanisms of DNA damage repair. Acylfulvenes (AFs) are a class of experimental anticancer agents that alkylate DNA in the minor groove, giving rise to N3-adenine adducts as a basis of inducing cytotoxicity in cancer cells. However, by efficiently stalling RNA polymerase, AF-DNA adducts activate transcription-coupled nucleotide excision repair (TC-NER), and removal of the AF adduct by TC-NER causes drug resistance. We aim to exploit the favorable therapeutic properties of AFs but reduce repair-associated resistance by creating compounds with the potential to induce DNA-protein binding interactions that promote cell death also in repair-proficient cells. Therefore, we designed new AF analogs that link various apolar and aromatic groups with the core sesquiterpene structure of AFs comprised of a fused (6,5) ring system and reactive cyclopropyl group. The effects of the compounds were characterized in human cancer cell lines that were either proficient or deficient in NER factors. The new analogs were cytotoxic to human cancer cell lines ranging from 0.02 – 107 µM concentration IC50 values, and repair-deficient cells were generally more sensitive. The results of this work indicate that chemical modification of AFs at the allylic alcohol position preserve the DNA-reactive nature of the compounds and suggests that stronger binding moieties towards the TC-NER repair proteins may promote cytotoxicity in repair-proficient cells.
About the Author
Laura Slappendel received her Master’s degree in Chemistry from the University of Groningen in 2020. Then she went for a PhD under the supervision of Prof. Shana J. Sturla at ETH to focus on targeting transcription-coupled repair for cancer therapy.
E Johanna L Stéen – 51
Amsterdam UMC
"Virtual screening of lead structures for P2Y12 receptor PET tracer development"
A PET tracer targeting the P2Y12 receptor would provide detailed information regarding neuroinflammatory conditions. To find novel lead structures with favorable properties for further development we have made use of a virtual screening approach.
Virtual screening of lead structures for P2Y12 receptor PET tracer development
Johanna L. Stéen1,2, Barbara Zarzycka3, Berend van der Wildt1,2 Danielle J. Vugts1,2, Iwan J.P. de Esch3 and Albert D. Windhorst1,2
Amsterdam UMC location Vrije Universiteit Amsterdam, dept Radiology & Nuclear Medicine, De Boelelaan 1117, 1081 HV Amsterdam, The Netherlands;
Amsterdam Neuroscience, Brain imaging, Amsterdam, The Netherlands
Division of Medicinal Chemistry, Amsterdam Institute of Molecular and Life Sciences (AIMMS), Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HZ, Amsterdam, The Netherlands.
The P2Y12 receptor (P2Y12R), a well-known drug target for antithrombotic agents, is also involved in microglia regulation during neuroinflammation. To get a detailed insight into P2Y12R-mediated dynamics in the brain, positron emission tomography (PET) can be applied. However, so far no successful brain permeable P2Y12 PET tracer has been reported.1 The aim of the present study is to find novel brain permeable lead structures, acting as P2Y12R antagonists by using a consensus virtual screening approach based on both ligand and structure-based modeling. To this end, complementary pharmacophore models were created by flexible alignment of six known P2Y12R antagonists and a high- throughput docking protocol was set up using a crystal structure of an antagonist-bound P2Y12R.2 After validation, the models were used in a screening of an in-house proprietary library (8427 drug-like compounds). The combined hit list from both approaches were processed (e.g., CNS PET MPO scoring3) and a set of chemically diverse hit representatives were tested in an in vitro binding assay. Hits with > 80% displacement were further explored by testing structural analogs, which in the end resulted in three new structural leads (Kiof 1–2 µM) for further optimization. On-going work focusses on optimization of these found leads.
References:
van der Wildt, B., et al. ACS Chem. Neurosci. 2021, 12, 23, 4465–4474;
2. Zhang K., et al. Nature 2014, 508 (7498), 115–118, (PDB code: 4NTJ, 2.62 Å);
Johanna Stéen works as a postdoctoral fellow with Bert Windhorst and Iwan de Esch in Amsterdam. Her research focusses on PET tracer development for targets in the central nervous system.
Omowaye Olaniyi Stephens Olaniyi Stephen Ola
Salem University Lokoja Nigeria
"EFFECT OF MORPHOMETRIC PARAMETER AND CLINICAL OUTCOME OF ETHANOLIC EXTRACT OF GARCINIA KOLA (BITTER KOLA) ON TRYPASONOMA PARAISTE IN WHITE ALBINO RATS"
EFFECT OF MORPHOMETRIC PARAMETER AND CLINICAL OUTCOME OF ETHANOLIC EXTRACT OF GARCINIA KOLA (BITTER KOLA) ON TRYPASONOMA PARAISTE IN WHITE ALBINO RATS
Pam Jerry a,b, Olaniyi Stephen Omowayea,DanielMakoloc, Peter FolorunshoAyodele,d* Emmanuel Sylvester Eneojoa
Department of Biosciences, College of Natural and Applied Sciences, Salem University, Lokoja, Nigeria
aDepartment of Biosciences, College of Natural and Applied Sciences, Salem University, Lokoja, Nigeria
dDepartment of Biochemistry, College of Natural and Applied Sciences, Lokoja, Nigeria
The Effect of Morphomeric Paramenter and Clinical Outcome of Ethanolic extract of Garcinia kolab (Bitter Kola ) on Trypanasoma congolense in White Albino Rats were evaluated. Eighteen (18) albino rats were used for this experiment. The ethanolic extract of Garcinia kola was found to possess a higher in-vitro activity, as it reduced the Trypanasoma congolense parasite within 5 minutes’ post incubation at concentrations 0.25, 0.5 and 1.0 mg/ml. This extract was therefore used to treat rats experimentally infected with Trypanasoma congolense at 100, 200, and 300 mg/kgbwt beginning 3 days’ post infection. At the termination of the experiment on day 7, the ethanolic extract significantly (P < 0.05) kept the parasite lower than what was observed in the infected-untreated rats. All the infected animals developed anaemia whose severity could be ameliorated by the extract treatment. Going by molecular docking result, the study showed that out of numerous compounds present in Garcinia kola, only four were potent compared with the standard drug. Therefore, it is suggested that Garcinia kola seed is a good therapeutic agent for Trypanasoma congolense.
Debela, E., Nigatu, K., Mirutse, G., Getachew, T. and Solomon, M.A.(2020).In vitro and in vivo Antitypasonoma activities of methanol extract of Echinops kebericho roots. Evidence-based complementary and alternative medicine. Evidence-Based complementary and Alternative medicine, 2022: 6.
Ekene, E.N. and Erhirhie, E.O. (2014). Garcinia kola: A review of its ethnomedicina-chemical and pharmacological properties. International Journal Current Research Review, 6(11): 1.
Emiru, A.Y., Makonnen, E., Regassa,F., RegassaF., and Tufa T.B. (2021). Antitrypasonomal activity of hydromethanol extract of leaves of cymbopogon citratus and sees of Lepidium sativum: invivo mice model. BMC Complementary Medicine and Therapies, 21:290
Getachew, A., Markos, T., and Yitagesu, T. (2019). Invivo and invitro anti-trypasonomal evaluation of crude methanolic extracts of crotalaria albicaulis and cistanche phelypaea against trypanosomaevansi. BMC Complementary and Alternative medicine, 9(4): 37-43
Habila, N., Abel, S.A., Zakari, L., Isaac. A.B., Emmanuel. H.,Monday. A.D., and Toafiq, O.A. (2010) evaluation of invitro activity of essential oils against trypasonoma brucei and trypasonoma evasi. Journal of parasitology Research, 2010:10
Isaac, C., Ohiolei, J.A., Ebhodaghe, F., Igbinosa, I.B., Eze, A.A. (2017). Animal African trypasonomiasis in Nigeria: A long way from elimination/eracdication. Acta Trop. 176: 323-331
Lori, P., Simon, C., Venessa,F., Mick, B., and Wendy, G. (2012). The life cycle of trypasonoma (nannomes) congolense in the tsetsefly. Parasites and vectors, 5:109
Magez, S., Caljon, G., Tran, T., Stilemans, B., Radwanska, M.A. (2010). Current status of vaccination against African trypanosomosis. Parasitology, 137:2017-27
Manˇourová , A., Leuner ,O., Tchoundjeu , Z., Damme, V.P., Verner, V., Prˇibyl., O.,and Lojka,.B. (2019). Medicinal potential, utilization and domestication status of bitter kola (Garcinia kola heckle) in west and central Africa. Forest, 10:124
Mesembe, O. E., Asuquo, O. R., Fischer, V. A., Udoaffah, G. U., Mfem, C. C and Kalu, N. N. (2013). Influence of Long-term Ingestion of Garcinia Kola Seed Diet on Sperm Count, Sperm Motility, and Fertility in the Wistar Rat. Journal of Health, Medicine and Nursing, 1.20-22.
Nagagi,Y.p; Temba, V; SilayoR.S; Kweka,E.J. (2018). Salient features of trypasonoma congolense in african animal trypasonomaisis in the sub-sahara africa. Vector-born disease and treatment.
Ng’wena, G.M., Odera, R., Ngiyawa, M.M., Mwaniki, B.M., and Okomu, W., (2020). Effects of allium sativum ethanolic extract on trypasonoma brucei brucei parasite morphometric parameters and clinical outcome in white albino laboratory rats. African Jounal of health science, 33(1): 1-13.
O’Boyle, N.M., Banck, M., James., C.A. (2011). Open Babel: an open chemical toolbox. Journalcheminform, 3:33.
Olivier, T.T., Martin, S., Armel, J.S., and Francis, N.T., (2016). Review on traditional uses, photochemical and pharmacological profiles of Garcinia kola heckle. Merit Research Journal ofMedicine and Medical Sciences, 4(11): 480-489
Sani, A., Umar, A.Z., Asmua, M., and Deepa, S. (2018). In vitro Study of antitrypasonomal activity of ethanolic leaf extract of Garcinia kola against trypasonoma brucei brucei. Asian Journal of pharmaceutical and clinical Research, 11(6): 417-419
Trott, O. and Olson, A.J. (2010). AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal comput chem, 31: 455-461.
Umar, I.A., Ibrahim, M.A., Fari, N.A., Esah, S., and Balogun, D.A. (2010). Invitro and invivo antitrypasonoma evansi activities of extracts from different pasts of khaya senegalensis. Journal of Cell and Animal Biology, 4(6): 91-95
Wanzala, E.N., Gikonyo, N.K., Murilla, G., Githua, M., and Hassanali, A. (2017). Invitro and Invivo anti trypasomoa activities of methanol extract of azadirachta indica stem-bark .African jounal traditionalComplement Alternative medicines, 14(6): 72-77
World health organisation. (2019). WHO interim guidelines for the treatment of gambiense human African trypanosomiasis.
https://www.who.int/trypanosomiasis_african/resources/9789241550567/en/ Web annex.com
Daniel Torres-Püschel – 52
Institute for Pharmaceutical Biology, University of Bonn
"Genome-mining- and metabolomics-guided discovery of novel antibiotics produced by Arabidopsis phyllosphere bacteria"
Genome-mining- and metabolomics-guided discovery of novel antibiotics produced by Arabidopsis phyllosphere bacteria
Daniel Torres-Püschel1, Aurélien Carlier2 Gabriele M. König1, Max Crüsemann1
Institute of Pharmaceutical Biology, University of Bonn, Bonn, Germany. 2: LIPME, University of Toulouse, Toulouse, France
Bacteria are well known for producing structurally diverse and biologically active specialized metabolites and they can be found in varied ecological niches. New environments shall be explored for further exploiting their yet untapped potential.
In a previous study, gnotobiotic Arabidopsis plants were inoculated with soil samples from the Gontrode forest, in Belgium. Hundreds of unknown biosynthetic gene clusters (BGCs) were discovered from 258 strains isolated from the rhizosphere and phyllosphere. This approach serves thus as an excellent study model for identifying novel natural products [1].
This project aims to investigate selected bacterial strains obtained by this strategy, which showed inhibitory activity against Acinetobacter baumanii and Staphylococcus aureus. Prioritization, identification, and prediction of natural products of interest (i.e., structural, biosynthetic novelty, bioactivity) from the BGCs thereof are being performed via “genome mining”, by employing state-of-the-art bioinformatic tools. For the expression of silent gene clusters in heterologous hosts, an advanced direct cloning strategy is utilized [2].
Here, research work conducted on novel non-ribosomal peptide synthetase (NRPS) BGCs from Paenibacillus algorifonticola and Streptomyces atratus strains is presented. A novel NRP was identified from P. algorifonticola by tandem mass spectrometry, and two silent NRPS BGCs from S. atratus were prioritized for cloning.
References:
Qi et al, Environ. Microbiol., 2021, 23(4), 2132–51.
"Substrate selectivity and inhibition of histidine JmjC hydroxylases MINA53 and NO66"
Substrate selectivity and inhibition of histidine JmjC hydroxylases MINA53 and NO66
Vildan A. Türkmen,1 Jordi C. J. Hintzen,1 Anthony Tumber,2 Laust Moesgaard,1 Eidarus Salah,2 Jacob Kongsted,1 Christopher J. Schofield,2 and Jasmin Mecinović1
Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense, Denmark. Department of Chemistry and the Ineos
Oxford Institute for Antimicrobial Research, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, OX1 3TA Oxford, United Kingdom.
Non-haem Fe(II) and 2-oxoglutarate (2OG) dependent oxygenases catalyse oxidation of multiple proteins in organisms ranging from bacteria to human. My research study focuses on the substrate selectivity and inhibition of the human ribosomal oxygenases (ROX) MINA53 and NO66, members of the Jumonji C (JmjC) 2OG oxygenase subfamily, which catalyse C-3 hydroxylation of histidine residues in Rpl27a and Rpl8, respectively. Assays with natural and unnatural histidine analogues incorporated into Rpl peptides provide evidence that MINA53 and NO66 have narrow substrate selectivities compared to some other human JmjC hydroxylases, including factor inhibiting HIF and JMJD6. Inhibition assay with Rpl peptides containing histidine analogues with acyclic side chains, including asparagine (Asn), glutamine (Gln) and homoGln, provide avenues for development of inhibitors selective for MINA53 and NO66.1
References:
Türkmen, V. A., et al., RSC Chem. Biol., 2023, in press
Zaheer Ul-Haq
Dr. Panjwani Center for Molecular Medicine and Drug Research, ICCBS, University of Karachi
"Broadening the Inhibitor Space by Utilizing Integrated In-silico Approaches; Dengue Virus Protease in View"
Broadening the Inhibitor Space by Utilizing Integrated In-silico Approaches; Dengue Virus Protease in View.
Mamona Mushtaq1, Sehrish Naz1, Sajda Ashraf1, Robert J. Doerksen2, Zaheer Ul-Haq1*
Dr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan.
Department of BioMolecular Sciences, University of Mississippi, Mississippi, USA.
Dengue is one of the leading arboviral infection imposing significant health burden on global population. The increased incidence and the threat of massive spread in near future has also been highlighted by the epidemiological studies. As per the recent records dengue is risking the lives of approximately 3.9 billion peoples globally1. Despite the massive economic burden and ever-growing global prevalence, definite therapeutic regimens are still not available to meet this pressing need. The dengue virus (DENV) protease (NS2B-NS3), a validated drug target is a two-component protein and is responsible for the processing of the translated polyprotein of the virus2. Since the active site inhibitors are subjected to off target effects, there is a growing interest in the development of allosteric inhibitors against DENV protease conferring greater selectivity3. In view of this, present investigation discusses computer assisted modelling of significant physico-chemical features required for the inhibition of DENV protease. At first computational mutagenesis was used to locate hot spot centers that are critical for the association of two sub units by means of energy profiles. In subsequent steps, an extensive structure based virtual screening (SBVS) was performed by utilizing the consensus docking, which yielded new inhibitors from the targeted small molecule databases. Further the stability dynamics and molecular mechanics were studied for the shortlisted hits with target enzyme in dynamic phase. In the following step binding free energies were calculated. Our results brought forth four compounds, which possess good binding affinities towards the target protein. We proposed that the identified small molecules might serve as novel scaffolds against NS2B-NS3 protease, that may lead towards the development of some potent anti-dengue therapeutics.
References
Patigo, A., Hengphasatporn, K., Cao, V., Paunrat, W., Vijara, N., Chokmahasarn, T., … & Khotavivattana, T. (2022). Design, synthesis, in vitro, in silico, and SAR studies of flavone analogs towards anti-dengue activity. Scientific Reports, 12(1), 1-18.
Götz, C., Hinze, G., Gellert, A., Maus, H., von Hammerstein, F., Hammerschmidt, S. J., … & Basché, T. (2021). Conformational dynamics of the dengue virus protease revealed by fluorescence correlation and single-molecule FRET studies. The Journal of Physical Chemistry B, 125(25), 6837-6846.
Jonniya, N. A., & Kar, P. (2022). Functional Loop Dynamics and Characterization of the Inactive State of the NS2B-NS3 Dengue Protease due to Allosteric Inhibitor Binding. Journal of Chemical Information and Modeling, 62(16), 3800-3813.
Lenci Karina Vázquez-Jiménez – 54
Centro de Biotecnología Genómica del Instituto Politécnico Nacional
"SEARCH FOR POTENTIAL INHIBITORS OF THE ENZYME TRIOSAPHOSPHATE ISOMERASE OF TRYPANOSOMA CRUZI BY VIRTUAL SCREENING"
Chagas disease is a neglected disease for which there is only nifurtimox and benzimidazole for its treatment, these cause serious adverse effects, which is why there is a need for new compounds with trypanocidal activity.
SEARCH FOR POTENTIAL INHIBITORS OF THE ENZYME TRIOSAPHOSPHATE ISOMERASE OF TRYPANOSOMA CRUZI BY VIRTUAL SCREENING
aCentro de Biotecnología Genómica del Instituto Politécnico Nacional, Reynosa, Tamaulipas, México
Glycolysis is the sole source of energy for Trypanosoma cruzi (T. cruzi) [1]. The enzyme triosephosphate isomerase (TIM) is involved in the fifth step of this pathway, and is a target for drug design [2]. On the other hand, the benzimidazole scaffold is an important structure in the development of antiprotozoal drugs [3]. Therefore, in this study a benzimidazole-based virtual screening was performed on the basis of ZINC15 data, the obtained compounds that complied with the Lipinski Rule were brought to molecular docking at the TIM interface of T. cruzi (TcTIM). Compounds with the most negative docking scores to the control ligand were selected as potential inhibitors of TcTIM. These compounds were grouped by interaction profile. The most representative compounds of each group according to the docking score were used to select compounds B2 and B5 (docking scores of -10.4 and -10.2 Kcal/mol, respectively) to be evaluated on the trypomastigote form of T. cruzi. In addition, they were analyzed by molecular docking on human TIM (HsTIM) to predict their possible selectivity. Compound B5 showed trypanocidal activity (IC50=179.71) better than benznidazole (IC50=191.28) and a low affinity for the HsTIM interface (-5.9 Kcal/mol) suggesting selectivity on TcTIM.
References:
Álvarez, G.; Aguirre-López, B.; Varela, J.; Cabrera, M.; Merlino, A.; López, G.V.; Lavaggi, M.L.; Porcal, W.; Di Maio, R.; González, M.; Cerecetto, H.; Cabrera, N.; Pérez-Montfort, R.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. “Massive screening yields novel and selective Trypanosoma cruzi triosephosphate isomerase dimer-interface-irreversible inhibitors with anti-trypanosomal activity”. Eur. J. Med. Chem., 45(12), 5767-5772, 2010.
Vázquez-Jiménez L. K.; Moreno-Herrera A.; A Juárez-Saldivar, Alonzo González-González, Eyra Ortiz-Pérez, Alma D Paz-González, Isidro Palos, Esther Ramírez-Moreno, Gildardo Rivera. “Recent Advances in the Development of Triose Phosphate Isomerase Inhibitors as Antiprotozoal Agents”. Current Medicinal Chemistry, 2021.
Aguilera, E.; Varela, J.; Birriel, E.; Serna, E.; Torres, S.; Yaluff, G.; de Bilbao, N.V.; Aguirre-López, B.; Cabrera, N.; Díaz Mazariegos, S.; de Gómez-Puyou, M.T.; Gómez-Puyou, A.; Pérez-Montfort, R.; Minini, L.; Merlino, A.; Cerecetto, H.; González, M.; Alvarez, G. Potent and selective inhibitors of Trypanosoma cruzi triosephosphate isomerase with concomitant inhibition of cruzipain: inhibition of parasite growth through multitarget activity. Chem Med Chem, 11(12), 1328-1338, 2016.
"Introducing an Active-Site Titrant of the SARS-CoV-2 Main Protease"
Introducing an Active-Site Titrant of the SARS-CoV-2 Main Protease
Rabea Voget, Julian Breidenbach, Alexandra Hingst, Christian Steinebach, Tobias Claff, Katharina Sylvester, Christa E. Müller, Michael Gütschow
Pharmaceutical Institute, University of Bonn, An der Immenburg 4, 53121 Bonn, Germany rabea.voget@uni-bonn.de
The exceptional medical and social situation resulting from the global COVID-19 pandemic makes research on SARS-CoV-2 a subject of high relevance. The main protease of the virus (Mpro) plays a critical role in viral replication and represents a particularly attractive drug target.1 This is underlined by the success of the already approved medication Paxlovid, which contains the Mpro-addressing drug Nirmatrelvir.2 The characterization of substrates and potential inhibitors of Mpro necessitates the knowledge not only of the protein concentration, but, more precisely, the concentration of the correctly folded, active enzyme. To meet this requirement, we developed an azatetrapeptide derivative as an active-site titrant for Mpro. The compound is susceptible to the nucleophilic attack of the active-site thiolate and forms a thioimidate with Mpro, resulting in an irreversible enzyme-titrant complex. We were able to illustrate this binding mode by cocrystallizing the titrant with Mpro. Quantitative inactivation of the enzyme by the titrant allowed us to determine the active-site concentration in various enzyme samples based on residual activities at defined titrant concentrations. We investigated different fluorogenic peptidomimetic substrates with respect to their solubility and their cleavage catalyzed by Mpro. For the kinetic analysis, the new active-site titrant was successfully applied.
References
Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An Updated Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2020, 2203, 1-29.
Lamb, Y. N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591.
Funded by the Volkswagen Foundation.
Antoine L. D. Wallabregue – 56
University of Oxford
"Development of bioreductive fluorescent probes to image different levels of hypoxia in human and plant cells"
Hypoxia sensing
Development of bioreductive fluorescent probes to image different levels of hypoxia in human and plant cells.
Antoine L. D. Wallabregue,1 Hannah Bolland,2 Shahneawz Khan,3 Emily Flashman,3 Ester M. Hammond,2 and Stuart J. Conway.1
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK.
Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Old Road Campus Research Building, Oxford, OX3 7DQ, UK.
Department of Biology, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK
Hypoxia (low oxygen levels) occurs in a range of biological contexts, including plants, bacterial biofilms, and solid tumors.1 It elicits responses from these biological systems that impact their survival, for example, hypoxia makes tumors more resistant to all forms of therapy and leads to poor patient prognosis.2 Hypoxia in the tumor microenvironment is heterogeneous (with O2 ranging from 0-6%) and its effects differ in an oxygen-dependent manner.3 Therefore, chemical imaging probes that enable the study of biological hypoxia are valuable tools to increase our understanding of disease-related conditions that involve low oxygen levels. Most existing small-molecule hypoxia-sensing images only very severe hypoxia (≤1% O2).4 Furthermore, commonly used antibody-based imaging tools for hypoxia are less convenient, as a secondary detection step involving immunostaining is required. Therefore, we have developed an efficient synthetic methodology that affords a palette of bioreductive fluorescent probes which are activated at a range of oxygen levels (0-6%) with emission maxima ranging through red, orange, and green. The deployment of these probes in human cell cultures,5 and for the first time in plants (roots and leaves), enables the imaging of different levels of hypoxia with unprecedented detail. These probes provide convenient alternatives to antibody-based imaging approaches.
References:
a) S. Akter et al., RSC Chem. Biol., 2021, 2, 1384, b) E. Loreti at al., Plants, 2020, 9, 745
a) Chen et al., J. Biomed. Sci. 2020, 27, 63
b) S. R. McKeown. Br J Radiol. 2014. 87, 20130676; c) J. C. Walsh et al., ARS. 2014, 21, 1516
a) S. L. Collins et al., ACS Chem. Biol. 2018, 13, 3354; b) K. Hanaoka et al., Chem. Commun. 2018, 54, 6939-6942; c) L. J. O’Connor et al., ACS Cent. Sci. 2017, 3, 20; d) R. B. P. Elmes Chem. Commun., 2016, 52, 8935; e) W. Piao et al., Angew. Chem. Int. Ed. 2013, 52, 13028.
Antoine joined the Conway group in 2020 as an EPSRC PDRA to develop chemical tools to image and target cellular redox components, particularly hypoxia. He aims to apply the tools in a range of biological settings including unusual ones (e.g., plants)
Antoine is also part of the redOx⇌KCL team, a collaborative program involving several research groups from the university of Oxford and King’s College London.
"Bayesian Inference Elucidates the Catalytic Competency of the SARS-CoV-2 Main Protease"
Literature of the SARS-CoV-2 main protease reveals considerable uncertainty regarding its enzymology, which impedes antiviral research. Using a Bayesian framework to sidestep limitations to traditional Michaelis–Menten analyses, we show the main protease is a more expedient enzyme than often appreciated.
Dimerization and Catalytic Efficiency of the SARS-CoV-2 Main Protease 3CLpro
Evans C. Wralstad, Jessica Sayers, and Ronald T. Raines*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
The main protease of SARS-CoV-2, 3CLpro, is a homodimeric enzyme indispensable to viral replication and presents an attractive opportunity for therapeutic intervention.1-4 Previously reported values of 3CLpro’s dimeric Kd and enzymic kcat/KM conflict by 106– and 103-fold, respectively.5–9 Establishing a confident benchmark of the intrinsic capabilities of this enzyme are essential toward combatting the ongoing pandemic. We characterize the dimerization and catalytic efficiency of authentic SARS-CoV-2 enzyme strictly by enzymology and using conditions optimal for catalysis. Specifically, we use the rigor of Bayesian inference in a Markov Chain Monte Carlo analysis of enzyme progress curves to circumvent the limitations of traditional Michaelis–Menten initial rate analysis.10 We report that SARS-CoV-2 3CLpro forms a dimer at pH 7.5 that has Kd = 16 ± 4 nM and is capable of catalysis with kcat = 9.9 ± 1.5 s−1, KM = 0.23 ± 0.01 mM, and kcat/Km= (4.3 ± 0.7) × 104 M−1s−1. Proteolysis is highly sensitive to disruption at increased solution ionic strength, which is largely a consequence of impaired dimerization. We conclude that 3CLpro is a more capable catalyst than appreciated previously, which bears implications regarding the design of protease inhibitors and other antiviral therapeutics targeting 3CLpro.
REFERENCES:
Grum-Tokars, V.; Ratia, K.; Begaye, A.; Baker, S. C.; Mesecar, A. D. Evaluating the 3C-like Protease Activity of SARS- Coronavirus: Recommendations for Standardized Assays for Drug Virus Res. 2008, 133 (1), 63–73.
Ferreira, J. C.; Rabeh, W. M. Biochemical and Biophysical Characterization of the Main Protease, 3-Chymotrypsin-like Protease (3CLpro) from the Novel Coronavirus SARS-CoV 2. Rep. 2020, 10 (1), 22200.
de Vries, M.; Mohamed, A. S.; Prescott, R. A.; Valero-Jimenez, A. M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J. C.; Ivanova, E.; Herrera, A.; Schinlever, A.; Loose, P.; Ruggles, K.; Koralov, S. B.; Anderson, A. S.; Binder, J.; Dittmann, M. A Comparative Analysis of SARS-CoV-2 Antivirals Characterizes 3CLpro Inhibitor PF-00835231 as a Potential New Treatment for COVID-19. Virol. 2021, 95 (10), e01819-20.
Morse, J. S.; Lalonde, T.; Xu, S.; Liu, W. R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-NCoV. ChemBioChem 2020, 21 (5), 730–738.
Chen, S.; Chen, L.; Tan, J.; Chen, J.; Du, L.; Sun, T.; Shen, J.; Chen, K.; Jiang, H.; Shen, X. Severe Acute Respiratory Syndrome Coronavirus 3C-like Proteinase N Terminus Is Indispensable for Proteolytic Activity but Not for Enzyme Dimerization. Biol. Chem. 2005, 280 (1), 164–173.
Hsu, M.-F.; Kuo, C.-J.; Chang, K.-T.; Chang, H.-C.; Chou, C.-C.; Ko, T.-P.; Shr, H.-L.; Chang, G.-G.; Wang, A. H.-J.; Liang, -H. Mechanism of the Maturation Process of SARS-CoV 3CL Protease. J. Biol. Chem. 2005, 280 (35), 31257–31266.
Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science (80-. ). 2020, 368 (6489), 409–412.
Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; Duan, Y.; Yu, J.; Wang, L.; Yang, K.; Liu, F.; Jiang, R.; Yang, X.; You, T.; Liu, X.; Yang, X.; Bai, F.; Liu, H.; Liu, X.; Guddat, L. W.; Xu, W.; Xiao, G.; Qin, C.; Shi, Z.; Jiang, H.; Rao, Z.; Yang, H. Structure of Mpro from COVID-19 Virus and Discovery of Its Inhibitors. Nature 2020, 582 (7811), 289–293.
Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-like Proteinase from SARS Coronavirus Catalyzes Substrate Hydrolysis by a General Base Mechanism. Biochemistry 2004, 43 (15), 4568–4574.
Hong, H.; Choi, B.; Kim, J. K. Beyond the Michaelis–Menten: Bayesian Inference for Enzyme Kinetic Analysis. In Computational Methods for Estimating the Kinetic Parameters of Biological Systems; Vanhaelen, Q., Ed.; Humana, New York, NY: New York, 2022; pp 47–64.
About the Author
Evans Wralstad is a fifth-year PhD candidate in the Raines lab at the Massachusetts Institute of Technology. He uses the principles of enzymology and protein chemistry to answer fundamental questions regarding human and viral proteins.
"Structural simplification of natural benzophenanthridines generates a candidate with highly selective inhibition of thioredoxin reductase for treatment of non-small cell lung cancer"
Structural simplification of natural benzophenanthridines generates a candidate with highly selective inhibition of thioredoxin reductase for treatment of non-small cell lung cancer
Qianhe Xu a, 1, Junmin Zhang a, b, 1, *, Pei Liu b, Zi-Long Song a, Yajun Chu a, Bingbing Chang a, Xiaojun Yao a, b, and Jianguo Fang c, a, *
a School of Pharmacy, State Key Laboratory of Applied Organic Chemistry, and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
b State Key Laboratory of Quality Research in Chinese Medicine, Dr. Neher’s Biophysics Laboratory for Innovative Drug Discovery, Macau University of Science and Technology, Macau (SAR), China
c School of Chemistry and Chemical Engineering, Nanjing University of Science & Technology, Nanjing, Jiangsu 210094, China.
* Corresponding authors: School of Pharmacy, State Key Laboratory of Applied Organic Chemistry, and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: zhangjunmin@lzu.edu.cn (J. Zhang).
School of Chemistry and Chemical Engineering, Nanjing University of Science & Technology, Nanjing, Jiangsu 210094, China; State Key Laboratory of Applied Organic Chemistry and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, China. Email: fangjg@njust.edu.cn or fangjg@lzu.edu.cn (J. Fang).
1 Authors are equal contributors.
Benzophenanthridine alkaloids, including sanguinarine, chelerythrine and nitidine, are natural products with a wide array of pharmacological activities. In this work, we simplify the structure of benzophenanthridines and obtain twelve three-membered phenanthridine alkaloids. The phenanthridine compound 6f shows improved potency in the specificity of thioredoxin reductase (TrxR, TXNRD) inhibition and cytotoxicity to the non-small cell lung cancer (NSCLC) A549 cells. TrxR is a selenoenzyme, whereas notably most TrxR inhibitors are susceptible to cross-reactions with biological thiols. Intriguingly, our results reveal that 6f selectively inhibits TrxR whereas with little reactivity to biological thiols, such as glutathione (GSH) and cysteine. Mechanism studies demonstrate that 6f also stimulates the production of reactive oxygen species in cells, which leads to the decrease of intracellular reduced thiols, the accumulation of oxidized glutathione (GSSG), and the decrease in the GSH/GSSG ratio. Furthermore, 6f causes cell apoptosis by inhibiting the intracellular TrxR to induce oxidative stress. Importantly, pronounced tumor regression is observed in nude mice bearing NSCLC following 6f treatment. The significant anticancer activity of 6f with well-defined action mechanism may provide a candidate for further development for NSCLC treatment.
"Phenolate-Induced N-O Bond Formation versus Tiemann-type Rearrangement for the Synthesis of 3-Aminobenzisoxazoles and 2-Aminobenzoxazoles"
We herein report the Phenolate-Induced N-O Bond Formation versus Tiemann-Type Rearrangement for the Synthesis of 3-Aminobenzisoxazoles and 2-Aminobenzoxazoles. While the selectivity for the two competing reactions was remarkably influenced by electronic and steric effects, TBACl was discovered as an effective additive, which promotes Tiemann-type rearrangement over N-O bond formation.
Phenolate-Induced N•O Bond Formation versus Tiemann-type Rearrangement for the Synthesis of 3•Aminobenzisoxazoles and 2•Aminobenzoxazoles
Hufnagel, B.;a,‡ Zhu, W. F.;a,‡ Franz, H. M.;a Proschak, E.; a,b,∥ Hernandez-Olmos, V.a,b,∥
a Institute of Pharmaceutical Chemistry, Goethe University Frankfurt, Max-von-Laue-Str. 9, 60438 Frankfurt am Main (Germany).
b Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Theodor-Stern-Kai 7, 60596 Frankfurt am Main (Germany).
‡ These authors contributed equally.
” These authors contributed equally.
Scaffold Hopping refers to the bioisosteric replacement of different core structures within the context of lead optimization.1 However, while peripheral functionalization is synthetically well established, seemingly small changes in the (hetero)cyclic scaffold often contradict with their retrosynthetic logic and each scaffold replacement requires establishment of its De–novo synthesis and functionalization chemistry.2 Synthetic precursors, which transform into multiple structurally similar heterocyclic products bear great potential for a synthetically more efficient access. With our ongoing interest in oxadiazolone-based annulation reactions, we herein report the Phenolate•Induced N•O Bond Formation versus Tiemann-Type Rearrangement for the Synthesis of 3•Aminobenzisoxazoles and 2•Aminobenzoxazoles.3 Starting from the same precursor (oxadiazolone), two heterocyclic isomers can be accessed under mild conditions. Substrate scope studies revealed that selectivity was remarkably influenced by electronic and steric effects. However, TBACl was discovered as an effective, safe, and affordable additive, which promotes Tiemann-type rearrangement over N•O bond formation allowing control of isomer formation.
References:
Sun, H.; Tawa, G.; Wallqvist, A. Classification of scaffold-hopping approaches. Drug discovery today 2012, 17 (7-8), 310–324. DOI: 1016/j.drudis.2011.10.024.
Woo, J.; Christian, A. H.; Burgess, S. A.; Jiang, Y.; Mansoor, U. F.; Levin, M. D. Scaffold hopping by net photochemical carbon deletion of azaarenes. Science 2022, 376 (6592), 527–532. DOI: 10.1126/science.abo4282.
Hufnagel, B.; Zhu, W. F.; Franz, H. M.; Proschak, E.; Hernandez-Olmos, V. Phenolate-Induced N-O Bond Formation versus TiemannType Rearrangement for the Synthesis of 3-Aminobenzisoxazoles and 2-Aminobenzoxazoles. ChemistryOpen 2022, 11 (12), e202200252. DOI: 10.1002/open.202200252.
About the Author
Wenxin Felix Zhu is a doctoral candidate at the research group of Prof. Proschak. With his ongoing interest in oxadiazolone-based annulations, he is happy to disclose part II of this research program at ACS Publications Symposium in Bonn.